Process Validation: The Essential Guide to Ensuring Product Quality and Compliance
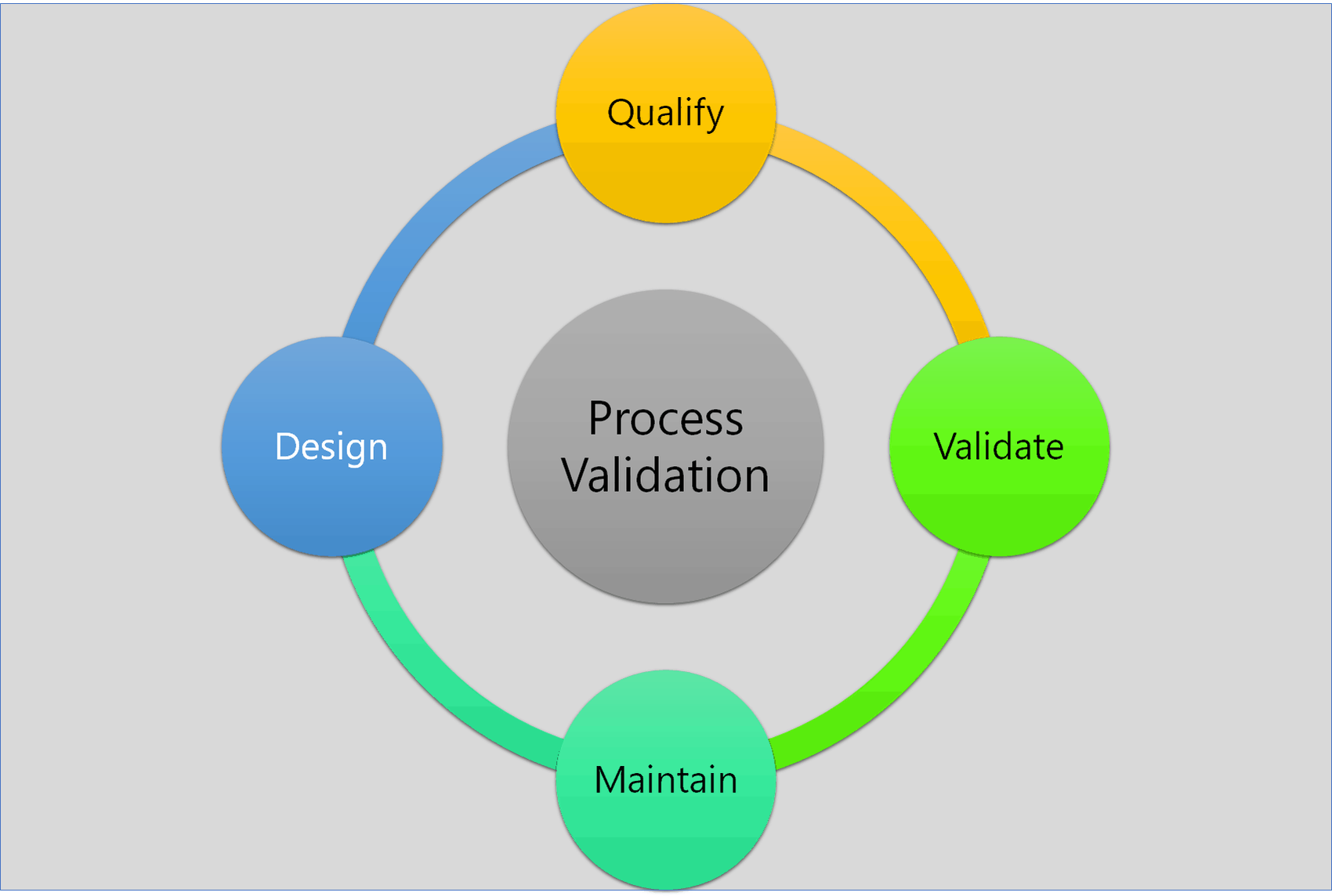
To assure product and patient’s safety, testing your product directly at the final stage is inadequate and may miss-out the other irregularities happened in the earlier stages of manufacturing.
So, instead of only final product, the whole manufacturing process must be tested for consistency in delivering the results. Right from the drug development stage through its discontinuation.
Hence, Process Validation is a requirement of:
- Current Good Manufacturing Practices (cGMP) Regulations for Finished Pharmaceuticals, 21 CFR Parts 210 and 211.
- Good Manufacturing Practice (GMP) Regulations for Medical Devices, 21 CFR Part 820, Quality System Regulation.
- Medical Devices – Quality Management Systems – Requirements for Regulatory Purposes, ISO 13485:2016.
Moreover, the FDA too now aligns the activities of process validation in pharma with the product life-cycle approach (explained further).
Table of Content
Overview
Drug quality depends on many important factors.
- Sound Product and Process Design
- Effective Process Control
- In-Process Quality Controls
- Final Product Testing
- Selection of Quality Components and Materials
- Destructive Testing (if applicable)
Additionally, the below quality aspects demonstrate the manufactured drug product is fit for its intended use.
- Product must meet its quality, safety, potency, and efficacy expectations.
- Quality built in every process step.
You might already know, the FDA defines process validation as:
The collection and evaluation of data, from the process design stage through commercial production which establishes scientific evidence that a process is capable of consistently delivering a quality product.
FDA Definition of Process Validation as per FDA
In short, process validation is a tool that addresses and measures the consistency of the drug manufacturing process.
If you were thinking what is the difference between Potency and Efficacy, here it is:
The amount of drug required to produce the desired effect is Potency, while Efficacy is the actual effect of a drug in therapeutic use.
Why is Process Validation required?
Process validation confirms “whether your process effectively controls the quality of your final product“.
It shows that your process consistently produces a quality product, batch to batch and product to product.
Here are some key reasons why process validation is important in life sciences:
- Ensuring Consistent Product Quality: By validating the manufacturing process, companies can ensure that the finished product consistently meets the desired quality specifications. This is essential for ensuring patient safety and meeting regulatory requirements.
- Compliance with Regulatory Requirements: Regulatory agencies, such as the FDA and EMA, require that companies validate their manufacturing processes in order to ensure that products are safe and effective for use. Failure to comply with these requirements can result in regulatory action, such as product recalls or fines.
- Cost Savings: Process validation can help identify potential issues early on in the manufacturing process, before they become larger and more expensive problems. This can help companies avoid costly rework, product recalls, and other negative outcomes.
- Improved Efficiency: Validating the manufacturing process can help identify opportunities for process improvement and optimization. This can lead to increased efficiency, reduced costs, and improved overall performance.
- Continuous Improvement: Process validation is an ongoing process, with continued monitoring and evaluation of the manufacturing process. This allows companies to continuously improve and optimize their processes over time, ensuring consistent product quality and patient safety.
A validation protocol, therefore, must include the requirements for test procedures and provision for data collection.
At least it should include:
- A number of process runs to ensure the process shows reproducibility and consistency. (Though this approach is now becoming an old way to validate the process and the FDA now recommends a product life-cycle approach that we’ll see further.)
- Bracketing for test parameters performed in such a way that upper and lower process limits are challenged to show the worst-case conditions.
- Evidence of material suitability.
- Performance and reliability of equipment and systems.
- Documenting and monitoring key process variables.
In-Process and Finished product testing are very important during process validation especially for tedious measurements of quality attributes and variables.
In these conditions, the qualification of each system communicating with other systems establishes a process validation approach.
All the validation-related activities must demonstrate Good Documentation Practices.
Approach to Process Validation
As per the product life-cycle approach, process validation activities are broken down into three stages.
| # | Stage | Activity |
|---|---|---|
| 1. | Process Design | Development and Scale-up |
| 2. | Process Qualification | Challenge Reproducibility |
| 3. | Continuous Process Verification | Periodic Verification |
Product and process development data are a prerequisite to securing a successful process validation.
Based on this data, a control philosophy is built to ensure that it achieves the desired product quality attributes.
Variations can severely affect product quality. So, it is important to:
- Identify the variations
- Analyze the magnitude of the variations
- Perform impact assessment
- Control the variations
- Build confidence in process performance
For already established product footprints, only stage 3 consideration is sufficient.
Now let’s understand the general aspects, recommended stages, and micro-activities in process validation.
General Aspects
For a successful process validation, proper planning and documentation of project activities are the key steps in the product life-cycle.
A sound scientific study that collects the product and process information helps to identify, monitor, and improve. Validating a process protects it from possible variations in order to produce the desired product consistently.
A risk-based approach is another aspect that deals with the critical quality attributes.
The term “critical” should show continuity in your decisions using a risk-based approach throughout the product lifecycle. Therefore, all such attributes should readily be available along with the well-plotted control measures.
Moreover, the principles of 3 stages and 4 types of process validation are quite correlative with each other.
3 Stages of Process Validation
- Process Design
- Process Qualification
- Continuous Process Verification
4 Types of Process Validation
- Prospective Validation
- Concurrent Validation
- Retrospective Validation
- Re-validation
Overall Scheme for Process Validation
Understanding an overall scheme makes process validation super simple. Let’s see how it is per the product life-cycle approach.
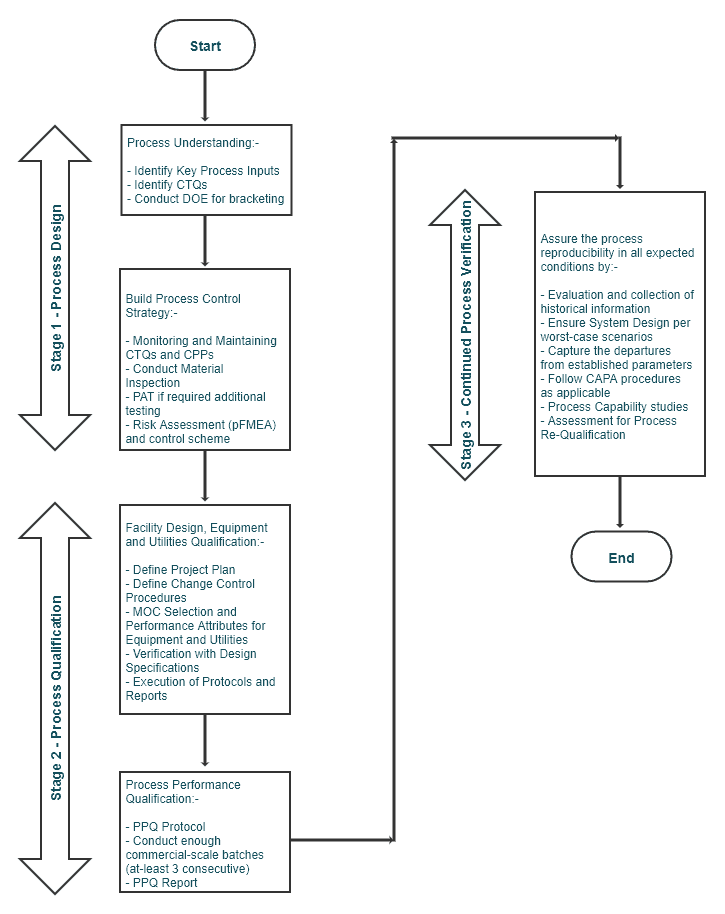
Stage 1 – Process Design (~Prospective Validation)
Process Understanding
This stage helps design a process suitable for routine commercial manufacturing that fulfills the pre-determined quality attributes.
Performing process development activities at this stage identifies key process inputs including the route of drug administration and critical quality attributes. Information collected through these activities supports the process design stage.
If the process variables are inadequate, lab- or exhibit-scale data may represent the overall variations for the time being.
Effective Process Design and Control relies on the knowledge and understanding gathered through the Design of Experiment (DOE) studies. This will build relationships between different process parameters and outputs.
Therefore, DOE helps industries to bracket their upper and lower process limits i.e., operating ranges. Process dynamics and simulation testing may help to identify potential risks before commercial manufacturing and avoid potential damage.
Importantly, documentation of all the above activities should reflect the rationale for your decision in process validation. For example, determining the number of temperature probes in a sterilization system and rationale for finalizing those numbers.
This then becomes a supporting document for further stages of process validation.
Building Process Control Strategy
It is not surprising that the variations in the input can damage the process output if not addressed at the right time. Therefore, it is important to build a control strategy that will ensure the process output remains unaffected.
This may include monitoring and maintaining critical process parameters and material attributes. One of the crucial aspects of identifying these parameters is to perform a proper risk assessment and mitigation plan.
While building up a control strategy, the process sometimes imposes limitations on the sampling and product uncertainties.
The FDA recommends conducting material inspections and equipment monitoring, especially using in-process monitoring and operational limits captured under respective BMRs.
For further analysis of materials and designing control loops, proper guides such as Process Analytical Technology (PAT) help optimize the process conditions for constant outputs.
This will ensure more confidence in process performance. However, it then requires a fresh approach to process qualifications as outlined in the FDA’s guidance, A Framework for Innovative Pharmaceutical Development, Manufacturing, and Quality Assurance.
A key stage in process validation of new products, Process Design activities are performed during the development stage and prior to commercial manufacturing.
This includes drawing up a schedule for trial plans, performing risk analysis, and establishing the required control strategy.
Summarizing Stage 1 – Process Design:
- Define the commercial production process
- Define CQAs and CPPs through pilot and exhibit batches
- Risk Assessment, pFMEA (process Failure Mode Effect Analysis)
- Plan controls to mitigate the risks
Stage 2 – Process Qualification (~Concurrent Validation)
At this stage, we’ve completed process design documentation showing a process that is capable of reproducing the desired results.
For further development, there are two critical aspects:
- Design of a Facility and Qualification of Equipment and Utilities
- Process Qualification
At this stage, the manufacturers should follow all the associated cGMP procedures.
The manufactured product will remain in quarantine until successful completion of this stage, after which the product can undergo commercial distribution if found acceptable.
1. Facility Design and Equipment and Utilities Qualification
Before moving on to Process Performance Qualification, assuring suitable facility design and commissioning as per Subpart C – Buildings and Facilities of 21 CFR Part 211 is a pre-requisite.
For equipment and utilities, common considerations are:
- Chalking out Project Plan with Change Control procedures, Qualification timelines, and Risk assessments.
- Appropriate selection of MOC for equipment and utility systems, including operation mechanism and performance attributes.
- Verification of equipment and utility systems built in accordance with the design specifications.
- Verification of equipment and utility systems operating at peak load consumption throughout their operating range.
- Documentation of activities with Protocols. Reports with clear conclusions addressing the required criteria outlined in the protocol and approved by the respective quality unit.
2. Process Performance Qualification (PPQ)
Successful Process Performance Qualification shows that the elements covered in the Process Design stage conform to actual scenarios. It also shows the commercial manufacturing process performs as intended.
Unless manufacturers comply with this, they can’t distribute the drug product commercially. Wherever possible, lab and pilot-scale data shall be used to support the commercial process for additional assurance.
Entering this phase shows that the process specifications have been established and equipment installation is deemed acceptable.
It is unnecessary to expedite the overall operating range if the process design data seems sufficient to assure performance.
Moreover, when a product or a process is not new, historical data analysis can be helpful, provided that the analysis is supported with sound statistical approaches and calculations (described further).
Heightened sampling and monitoring plans should also be on the checklist as and when necessary to confirm uniform product quality.
Reusing process consumables such as filters or resins should show through relevant lab studies that they don’t degrade the product quality and integrity. If this is the case, the PPQ protocol should cover this approach too.
2.1) PPQ Protocol
PPQ Protocol is a written document covering the following specifications hierarchically.
- Process control
- Process description
- Sound process limits
- Control methods
- Impact assessment
- Personnel training and qualifications
- Manufacturing conditions like:
- Raw materials
- Operating ranges
- Process limits
- Testing instructions and acceptance criteria for:
- Process characterization
- Sound sampling plan
- No. and location of sampling points
- No. of samples
- Sampling frequency
- In-process testing
- Final testing
- Process indicators
- Process variability
- Deviation handling and CAPA procedure as per applicable SOP
- Non-conformance management
- Expected outcome
- Productivity
- Yield
- Loss
2.2) Protocol Execution
Upon review and approval from all stakeholders, the protocol execution begins as per respective BMR and SOPs under normal operating conditions.
Rationale and Quality Unit approval is required before concluding all the observed deviations during execution.
2.3) PPQ Report
Report preparation can begin contemporaneously unless otherwise justifiable, documenting and examining the outcomes with PPQ protocol.
Report content should include at least the following things.
- Summary and analysis of the data collected during the execution, including non-conformances
- Missing data from the protocol, if any, should be part of the report
- Unexpected observations should also get tested in the report
- Review all protocol references
- Discuss and summarize the observed deviations such as failed test results and OOS. This should also propose a CAPA strategy to either modify the process or controls.
- An explicit statement of PASS or FAIL based on the requirements outlined in the PPQ protocol. Otherwise, the report should mention the expected conditions to be considered PASS or FAIL with suitable reference.
Once all the above aspects are fulfilled, the report can then route for review and approval of all the stakeholders.
Process Qualification activities are performed to demonstrate and assure that the process will remain in a state of control during commercial manufacturing.
Stages 1 and 2 should involve measurement systems for the identified attribute/s and subsequent control loops.
Summarizing the Stage 2 – Process Qualification:
- Conduct enough commercial-scale production batches consecutively to qualify the process that was designed in Stage-1
- Closely monitor all the process parameters
- Detail out the collected data for assessing CQAs and CPPs for consistency and variabilities
- Design the facility
- Perform equipment and process performance qualification
Stage 3 – Continued Process Verification (~Retrospective Validation)
This stage aims to safeguard the reproducibility of process performance in all expected conditions. To achieve this, ensuring system design as per worst-case scenarios is the key.
This means the system should detect any unplanned deviations from established parameters from previous stages.
Apart from system design, cGMP aspects such as collection and evaluation of process performance-related data helps the firms:
- To detect and address the variations.
- Implement CAPA and control quality attributes appropriately throughout the process. This data may include trends, charts, material inspection reports, etc.
Companies can also use statistical tools like process capability studies to ensure the process remains stable and consistent.
The use of quantitative and statistical tools allows assessment of both intra-batch and inter-batch variations and confirms the continued process verification approach. These estimates helps in periodic adjustments to establish heightened sampling and monitoring.
Additional checks to detect the process variations are:
- Out-of-Specification (OOS) checks
- Market complaints
- Yield fluctuations
- Deviation reports
- BMR
- Analyzing raw material inspection reports
- Feedbacks
- IPQA inspections
- Preventive maintenance and calibration schedules
- Re-qualification based on periodic assessment
This data helps to optimize the process by adjusting and documenting the operations with suitable rationale. In short, this ensures continued process verification.
Summarizing Stage 3 – Continued Process Verification:
- Qualify the process by evaluating and collecting historical information of previous batches and analyze how well process parameters remain in the acceptable range.
- Perform periodic analysis of data to conclude the requirement of re-qualification of process.
Type 1 – Prospective Validation
Prospective Validation is conducted before the distribution of a new product. Prospective Validation and Stage 1 – Process Design mostly includes considerations that deal with an altogether new product introduction.
Therefore, it becomes a completely planned program to design and validate a process, including a fundamental document like Validation Master Plan (VMP) and Strategy Protocol.
The VMP may include the following:
- Planning and Scheduling
- Summary and current status of the facility, systems, equipment, and/or process
- Change control procedures required to follow
- Reference to this VMP to execute the validation activities
Elements of Prospective Validation includes: Equipment and Process qualifications like Installation Qualification, Process Performance Qualification, Product Performance Qualification, and associated documentation.
Equally, assessment of revalidation becomes important for the timely validated state of product and process. Remember, the aim is to establish documented evidence before commercial manufacturing that the process will perform as intended.
Type 2 – Concurrent Validation
Unlike Prospective Validation, Concurrent Validation occurs parallel to product manufacturing.
This means companies can distribute the batches commercially before the approval of validation activities provided that the product must meet pre-defined quality specifications. By then, it should be quarantined.
The rationale for this should be documented with supporting data used in making a decision.
Although all documentation requirements are as per Prospective Validation, this approach is decided based on product or market limitations like short shelf-life, high demand short supply, etc.
The involvement of all stakeholders in the review and approval of concurrent validation, including regulatory bodies, is recommended because the FDA considers this method of validation a rare condition.
Assessment and conclusion of performing this type of process validation should be documented with justification.
The FDA outlines several expectations for the concurrent release of PPQ batches:
- Prior to commercial distribution of the PPQ batch, expect to prepare a dedicated protocol mentioning this special case.
- Potential situations and rationale should be part of the PPQ protocol. Also, the complete execution of the PPQ protocol and data evaluation is a must before drawing any conclusions.
- Additional process design and qualification become necessary when Stage 2 – Process Qualification fails.
- A lot released concurrently must comply with cGMP and other regulatory requirements despite the pending clearance on PPQ batches. Also, it must pass all the acceptance criteria outlined in the PPQ protocol.
- Firms can release a lot when they meet the required confidence levels for quality attributes applicable to that product.
- A fast-track assessment of market complaints and feedback is expected when a lot is released concurrently.
Type 3 – Retrospective Validation
Retrospective Validation is based on historic control, testing, and production data for a product that is already in commercial distribution.
Performing this validation does not demand special requirements as such. This type of process validation may or may not require the firms to reperform all the validation activities previously executed.
Sometimes, a conclusive summary of not performing validation may be provided showing how the system works in its desired state flawlessly since commissioned.
In some cases, validating only specific parts would bring confidence to the process performance. To support such a decision, data and statistics should be attached.
As the system is already set up and running, Design Qualification is deemed unnecessary for retrospective validation whereas all the other documentation activities remain identical to the Prospective and Concurrent Validation.
Type 4 – Re-Validation
Re-validating the existing process becomes essential, especially when any changes are made in the process or its environment, including changes in:
- Raw material
- Raw material supplier
- Product formulation
- Packaging material MOC
- Packaging material supplier
- Process
- BMR step modifications
- Changes outside the established process range
- Transfer from one site to another
- Equipment
- Relocation (Major change)
- Removal
- Major modifications (Through proper assessment)
- Product changeover
- Software upgrade
- Changes in the area (HVAC Changes)
Re-validation should therefore start based on established procedures outlining such specific instances. The extent of this re-validation depends on the nature of the change and the potential impact on product quality assured in previous validation stages.
This means, for example, a material change in product packaging would not require complete process validation, and focusing on packing operation would suffice the requirement.
However, it becomes essential to determine the start and endpoint and the subsequent effects of re-validation based on the nature of change. Documenting the change control will establish the extent of such re-validation.
V Model for Validation
Another term we generally come across is V-Model for validation. This is more of a simple presentation than a requirement to promote an easy understanding of the overall validation cycle.
In 1994, GAMP 1st Edition, ISPE introduced this model which shows how and where a validation phase starts and ends, including the correlation between them.
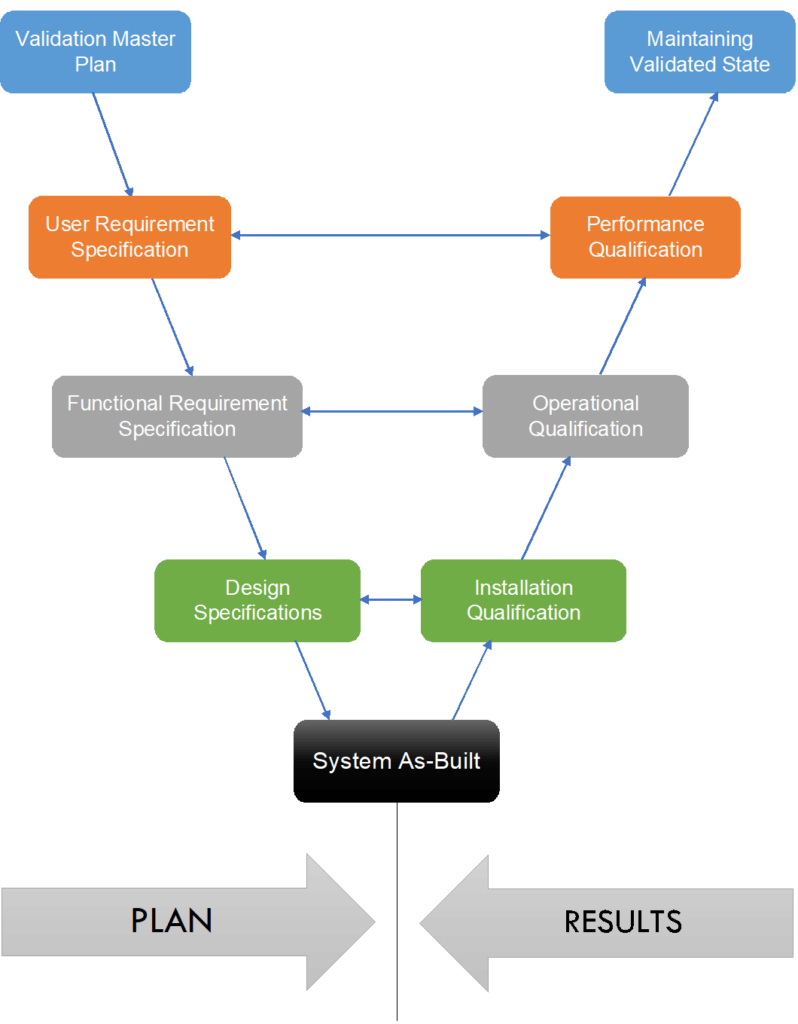
The left arm of V represents the planning stage or specification phase, while the right arm of V represents the obtained results. The point of convergence represents the system establishment.
- 1st correlation (Green) shows that the system is installed as per design specification.
- 2nd correlation (Gray) shows that the system operates as per functional specifications.
- 3rd correlation (Orange) shows that the system performs as per user requirement specifications.
- The starting point of the V-Model is Validation Master Plan and the endpoint is maintaining a validated state.
The V-Model exhibits a reasonable pattern that assists to order the activities defining project scope and execution.
Validation Master Plan (VMP)
The Process Validation Master Plan summarizes the overall validation strategy across the organization containing documentation for validation of life-cycle elements from commissioning to decommissioning.
These elements include Process, Equipment, Utilities, Cleaning Procedures, Computer Systems, Test Methods, etc. VMP also captures the validation status of each component to evaluate the impacts on product quality, efficacy, or even identity.
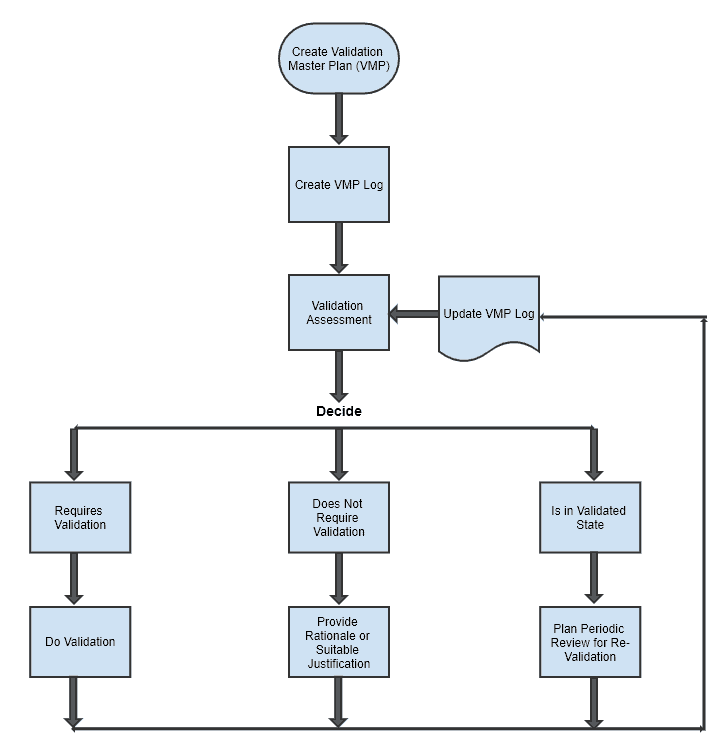
VMP Log
This is a control document with a list of reference numbers related to all validation activities including protocols, reports, and other important information.
This log is maintained to support the validation status of each applicable and non-applicable component.
The following items are considered to be part of VMP at least:
- Product Name
- Validation Reference Number
- System Details
- System ID
- Review Date
- Re-validation Date (if required)
- Deviation and Non-Conformance Reference Numbers (if applicable)
VMP Process Flow
Below mentioned is a simple process flow chart for different activities covered under VMP.
VMP Components
The following components are captured under the Validation Master Plan.
- Drug Products
- Manufacturing Process
- Utilities in direct contact with the product or process
- Validation Strategies and Plans
- Validation Protocols and Reports
- Relevant SOPs
- Roles and Responsibilities
- Flow of planned activities
- Equipment and ancillary systems that must be validated
- Periodic Review Program
- Other components dealt with validation
VMP must address the following aspects to ensure its completeness, accuracy, and frequency.
- Changes in:
- Process
- Product
- Equipment
- Ancillary Systems
- Responsible Functions
- Validation Plans, Protocols, and Reports
- Any additional validations other than proposed in strategies
- Updating VMP logs for any addition, removal, or modification of validation components
Periodic Reviews
The purpose of performing periodic reviews deals with the concern – “whether the cumulative changes have affected the manufacturing process performance“. Supporting data to conclude periodic reviews can be collected from various sources such as:
- BMR
- Environmental Monitoring
- Test Reports
- Trends
- Deviations
- CAPA
- Failures
- Market Complaints
- Calibration Records
- Preventive Maintenance Records
- Quality Audit Records
- Process Monitoring and Control
Once all the events are gathered and ready, periodic reviews shall be documented as per applicable templates to deem the component status as validated OR non-validated OR validation not required with ready justifications.
At the same time, update the VMP log for the made decisions. This way the firm can perform effectively in terms of managing process validation cycles.
FAQs: Process Validation Life-Cycle Documents
What is a Validation Plan or Strategy?
This document outlines the scope of activities at the very beginning of the process validation and is considered one of the main components of the process validation life-cycle.
As the project progresses, the validation plan may need revisions for any potential changes either in scope or the project itself.
This should mention the other validation-related components that are applicable such as DQ, IQ, OQ, PQ, PV (Process Verification), etc., including worst-case scenarios and the deviations.
What is User Requirement Specification (URS)?
URS includes all the requirements that the process or system must meet. Either solely created by the user or with the help of a vendor with approval from Quality. The various types of requirements specified in this document include:
– Automation Requirements such as alarms, interlocks, and process control
– Safety requirements such as fail-safe conditions, safety-interlocks
– Electrical Requirements
– Regulatory Requirements
– Process Flow Chart
– Technical Requirements
– System Component Details
– Preferred suppliers
– Documentation
What is Functional Requirement Specification (FRS)?
This document involves the requirements relating to the functional aspects of the equipment or system in a way that fulfills the URS goals. Therefore, this document is considered as a counter to the URS.
As the name suggests, this document is generally prepared by a vendor or supplier. As per V-Model, the functional specifications correspond to the operational qualification by testing each of the parameters.
What is Design Specification (DS) OR Technical Specification (TS)?
This document should include basic design and engineering requirements to facilitate the system installation. Highlighting technical specifications may involve features countering the URS. This may include:
– Design data like Pressure, Temperature, Volume, etc.
– MOC data
– Process related features
– Instrument lists
– Utility Provisions
– Safety Provisions
– Process Control and extent of automation
What is Factory Acceptance Test (FAT)?
Tests are carried out in the factory where the system is constructed termed as FAT. The objective of this testing is to ensure that the system functions as per its operational requirements laid down in URS.
A cross-functional team from the user side will visit the factory to witness the same. The vendor then demonstrates the functionality of each component in scope and prepares the report summarizing the activities. The user then agrees or disagrees and decides on a further action plan.
Note: FAT is considered optional in the validation life-cycle approach as it depends on mutual understanding rather than a mandatory thing.
– Identify and rectify potential defects before the system is transferred to the user site.
– Use as a basis for user acceptance at the vendor level.
What is Design Qualification (DQ)?
The objective of Design Qualification is to provide documentary evidence to show that all the aspects of URS for Regulatory, cGMP, Safety guidelines are considered in designing the system.
– Some of the DQ components include
– Process and Instrumentation Diagram (P&ID)
– General Arrangement (GA) Drawing
– 3-Dimensional Scheme (Optional)
– System Architecture
– Electrical Drawings
– Bill of Quantity (BOQ)
– Utility details
– Calibration requirements
What is Quality Risk Management (QRM)?
This document should identify the following
– Potential risk factors
– Risk Elements
– The probable cause of the risk element
– Risk Classification
– Risk Control or Mitigation through CAPA/Change Control/IQ/OQ/PQ
– Controlled Risk Classification
– Risk Acceptance
The QRM is considered one of the core aspects of process validation, according to FDA.
What is Installation Qualification (IQ)?
Establishing confidence through documented evidence that “the system and ancillary components have been installed correctly and are capable of consistently operating within established limits.”
Following is the list of activities considered during Installation Qualification (IQ)
– Document Verification
– Verification of all the drawings approved during DQ
– Weld joint inspection
– Slope verification
– BOQ verification
– Input-Output Testing (I/O) if applicable
– Pressure Hold Testing
– Material finish verification (radiography)
– Cleanroom Verification (Temperature and Relative Humidity)
What is Operational Qualification (OQ)?
Established through documented objective evidence that “the system operates and controls in accordance with Functional Requirement Specifications and addresses the operating principles outlined in URS”.
Following activities considered during an Operation Qualification (OQ) stage
– System Start-up and basic functionality checks
– In-depth functionality testing
– Alarm and Interlock verification
– Power failure testing
– Spray-ball coverage test (Cleaning verification)
Besides these, more process-specific operations may be conducted to establish documented evidence such as working volume verification, safety valves, etc.
What is Computer System Validation (CSV)?
Consider this as a software validation that follows the same V-Model we discussed in this article. CSV establishes through documented objective evidence that the computer system fulfills the pre-defined software requirement specifications.
CSV is considered through FDA, EU, ICH, and even WHO’s perspective for the following domains:
– Pharmaceutical Companies
– Biotech Companies
– Medical Device Manufacturers
– Storage and Distribution Companies related to healthcare products
CSV is applicable to GxP systems directly or indirectly supporting process automation and is also a part of process validation.
Components of CSV include
– Computer Installation Qualification (CIQ)
– Computer Operational Qualification (COQ)
What is Performance Qualification (PQ)?
Performance Qualification (PQ) is established through documented evidence that a process consistently produces a product meeting its pre-determined specifications under all foreseen conditions.
PQ is a process that continuously demonstrates the system performance in accordance with the commercial process specifications outlined in URS.
What is Process Verification (PV)?
There is a lot of confusion between process verification and validation. The FDA defines process verification as “confirmation by examination and provision of objective evidence that specified requirements have been fulfilled.”
Process Verification (PV) follows a system-centric approach while Process Validation follows User Requirement Specifications. This means Process Verification confirms whether specifications are being met according to preset parameters and Process Validation according to the user.
What is the Validation Completion Report?
A report summarizing all the aspects outlined in the Process Validation Plan along with the following observations
– Deviations
– Non-Conformances
– Process Departures
– In-Process Testing Results
– Finished product test results
– Clear Statement of Meeting or Not Meeting the Goals
– Future action plans
The report should also document all the deliverables expected and outlined in the planning stage.
Useful Tools In Manufacturing
Apart from regular manufacturing activities, the following tools are also used to improve the effectiveness of process validation. These tools help in monitoring and optimizing the process variables ultimately effecting productivity.
- Lean and 6 Sigma
- pFMEA
- Process Capability
- 5 Why Analysis
- Fault Tree Analysis
- Fish Bone Diagram / Ishikawa Diagram / Cause and Effect Diagram
- Quality by Design (QbD)
- Design of Experiments (DOE)
- Process Analytical Technology (PAT)
- Gage R&R Study
These tools are explained in detail here: Quality by Design and 9 Allied Tools To Thrive Productivity.
Conclusion
Whether you start from scratch or re-validate your existing processes, in process validation, every step shall be connected with a rationale.
It is management’s responsibility to maintain a scientifically sound strategy.
Purpose of this post is to put process validation in a single picture rather than understanding different pieces separately. Hope this effort somehow made an impact.
How do you conduct process validation in your manufacturing facility? Do these aspects are enough or which additional efforts do you put? Either way, let me know in the quick comment below.
Such a thorough article! This has covered almost everything about process validation. Thank you for this quality content 👌
This article really helped me to crack my interview. Really thanks for this superb and one page information of such a broad concept. I will surely follow this site hereafter and would recommend other professionals for easy and in-depth understanding.
Congratulations Mr. Sinha M. K.! Stay tuned for more value addition. Wishing you all the success.