FDA’s 21 CFR Part 11: The Definitive Guide

As per different stats in the last 5 years, “Out of every 10 FDA warnings (Form 483), 8 were data integrity gaps”. Meaning, nearly 80% of the warning letters had data integrity issues. A large chunk of them were related to digitally generated records.
There are two basic reasons that result in 21 CFR Part 11 non-compliance:
- Mis-interpretations
- Inadequate implementation
Since its origin back in March 1997, the FDA’s 21 CFR Part 11 has caused great confusion among healthcare providers who wanted to use electronic systems to scale up their business.
To address this, the FDA released a guidance document in August 2003 clarifying the scope and application of this regulation. The FDA says this guidance document is not the revision of the actual law, instead, their current thinking on the topic and a narrow interepretation.
Audit trails, Signatures, Validation, Record management, etc., are of course there. But 21 CFR Part 11 is much more than that.
In this detailed guide, you’ll learn:
- Structure of 21 CFR Part 11
- How CFR documentation is organized
- How to interpret and comply with Part 11;
If you are ready to go “all in” with 21 CFR Part 11, this guide is for you.
Part 11 is not applicable if the system –
- Either generates paper printouts/records or
- If it was commissioned before August 20, 1997.
Table of Content
What is 21 CFR Part 11?
21 CFR Part 11 is a regulatory framework designed by the FDA to guide drug makers on how to create, manage, and maintain electronic records and electronic signatures.
It also says in what conditions drug makers become eligible for compliance with this regulation.
The term comes from Title 21 of the Code of Federal Regulations (CFR). In effect since January 2007, 21 CFR Part 11 governs electronic records and signatures in regulated industries like pharmaceuticals, biotech, medical devices, etc.
Totally, the CFR has 50 titles.
Out of them, Title 21 deals with food and drugs.
Again, Title 21 has 3 chapters.
- Chapter I – Food and Drug Administration
- Chapter II – Drug Enforcement Administration
- Chapter III – Office of National Drug Control Policy
To understand this sequence easily, check out the below table.
Sequence (Swipe right while on mobile screen)
| Title | Code | Chapter | Subchapter | Part | Subpart | Sections |
| 21 | CFR | I | A – General | 11 | A, B, C | 11.1, 11.2,… |
Terminology
| Term | Meaning |
|---|---|
| 21 | Title that deals with food and drugs. |
| CFR | Code of Federal Regulation |
| Chapter I | Guidelines related to the Food and Drug Administration (FDA) basically drawn from the Food, Drug, and Cosmetic Act, 1938 |
| Subchapter A – General | Chapter I includes Subchapters named A, B, C and so on. Part 11 is covered under Subchapter A – General. |
| Part 11 | Regulations related to electronic records and electronic signatures. |
Through this regulation, Electronic Records and Electronic Signatures became as valid as paper records and handwritten signatures.
However, the 21 CFR Part 11 does not mandate its use. In fact, it guides the organizations that choose to use digital systems for compliance.
Part 11 is divided into 3 subparts:
A) General Provisions
B) Electronic Records
C) Electronic Signatures
21 CFR Part 11 Compliance Checklist
Here is an overall breakdown of 21 CFR Part 11 (Zoom or open the image in a new tab).
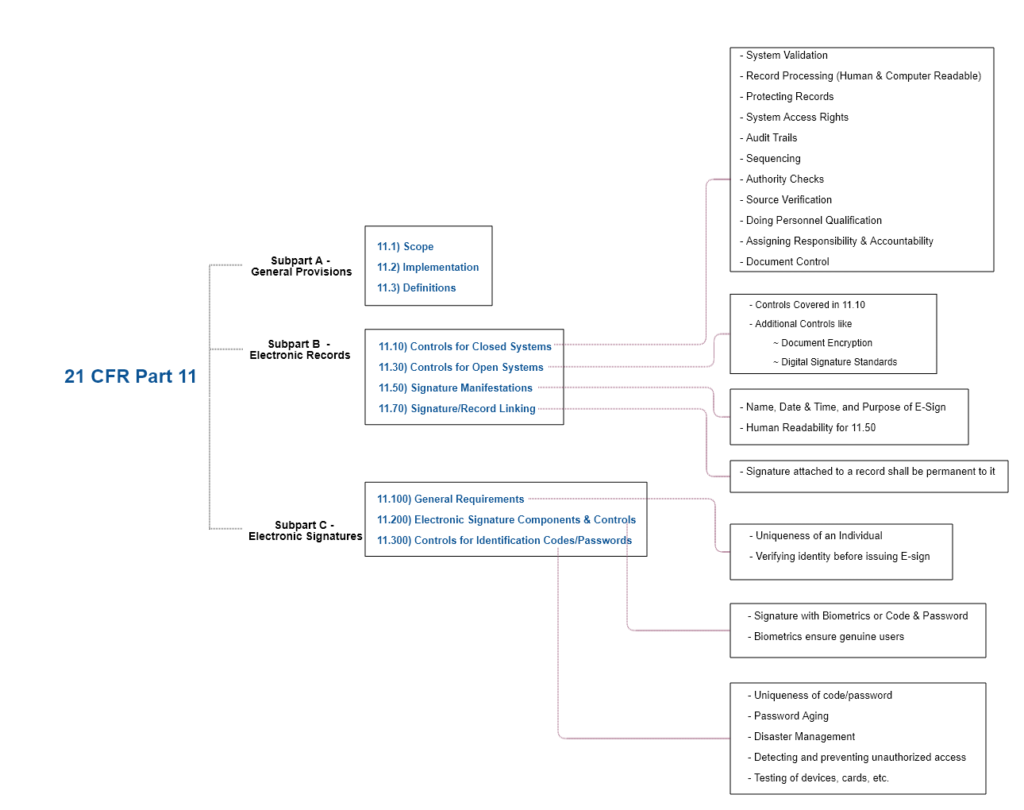
So, use the systems that meet every component specified in the 21 CFR Part 11 Compliance Checklist above and as we dive right in step-by-step.
Subpart A – General Provisions
11.1 – Scope
| Regulation | Interpretation |
|---|---|
| (a) The regulations in this part set forth the criteria under which the agency considers electronic records, electronic signatures, and handwritten signatures executed to electronic records to be trustworthy, reliable, and generally equivalent to paper records and handwritten signatures executed on paper. | The FDA wants to recognize electronic records and signatures AS VALID AS paper records. So, Part 11 talks about that perspective. |
| (b) This part applies to records in electronic form that are created, modified, maintained, archived, retrieved, or transmitted, under any records requirements set forth in agency regulations. This part also applies to electronic records submitted to the agency under requirements of the Federal Food, Drug, and Cosmetic Act and the Public Health Service Act, even if such records are not specifically identified in agency regulations. However, this part does not apply to paper records that are, or have been, transmitted by electronic means. | Each electronic record used in a regulated process needs to follow Part 11. It includes business e-mails too (except for the paper records in the form of e-mail attachments). |
| (c) Where electronic signatures and their associated electronic records meet the requirements of this part, the agency will consider the electronic signatures to be equivalent to full handwritten signatures, initials, and other general signings as required by agency regulations, unless specifically excepted by regulation(s) effective on or after August 20, 1997. | If E-signs meet the Part 11 requirements, they can replace paper signatures. But some regulations may specifically demand ink signatures. |
| (d) Electronic records that meet the requirements of this part may be used in lieu of paper records in accordance with 11.2, unless paper records are specifically required. | If E-records meet the Part 11 requirements, they can replace paper records. But some regulations may specifically demand paper records. |
| (e) Computer systems (including hardware and software), controls and allied documentation maintained under this part shall be readily available for, and subject to, FDA inspection. | Firms should document the rationale for taking decisions under each regulation of this section. |
11.2 – Implementation
| Regulation | Interpretation |
|---|---|
| (a) For records required to be maintained but not submitted to the agency, persons may use electronic records in lieu of paper records or electronic signatures in lieu of traditional signatures, in whole or in part, provided that the requirements of this part are met. | As long as electronic records satisfy Part 11, a company can use it with/without paper records even when the regulated records submission to FDA is pending. |
| (b) For records submitted to the agency, persons may use electronic records in lieu of paper records or electronic signatures in lieu of traditional signatures, in whole or in part, provided that: (1) The requirements of this part are met; and (2) The document or parts of a document to be submitted have been identified in public docket No. 92S-0251 as being the type of submission the agency accepts in electronic form. This docket will identify specifically what types of documents or parts of documents are acceptable for submission in electronic form without paper records and the agency receiving unit(s) (e.g., specific center, office, division, branch) to which such submissions may be made. Documents to agency receiving unit(s) not specified in the public docket will not be considered as official if they are submitted in electronic form; paper forms of such documents will be considered as official and must accompany any electronic records. Persons are expected to consult with the intended agency receiving unit for details on how (e.g., method of transmission, media, file formats, and technical protocols) and whether to proceed with the electronic submission. | In public docket no. 92S-0251, the FDA lists the types of e-records they accept. If in doubt, contact the receiving unit at the FDA before submitting e-records. Once records are submitted to the FDA, companies can use e-records on the condition that the records comply with Part 11 requirements. |
11.3 – Definitions
| Regulation | Interpretation |
|---|---|
| (a) The definitions and interpretations of terms contained in section 201 of the act apply to those terms when used in this part. | Section 201, Food, Drug and Cosmetic Act already includes definitions. So, they carry the same meaning in Part 11 if used. |
| (b) The following definitions of terms also apply to this part: | Definitions of the terms in the Food, Drug, and Cosmetic Act that apply to Part 11 are: |
| (1) Act means the Federal Food, Drug, and Cosmetic Act (secs. 201-903 (21 U.S.C. 321-393)). | Act – Food, Drug, and Cosmetic Act |
| (2) Agency means the Food and Drug Administration. | Agency – Food and Drug Administration |
| (3) Biometrics means a method of verifying an individual’s identity based on measurement of the individual’s physical feature(s) or repeatable action(s) where those features and/or actions are both unique to that individual and measurable. | Biometrics – Method of physical identity verification. For example, recognition through Face, Fingerprint, Iris, Voice, etc. |
| (4) Closed system means an environment in which system access is controlled by persons who are responsible for the content of electronic records that are on the system. | Closed System – Persons who control the records of the system, control the access to it. Meaning, the organization that generates the records, stores the record. So, the record is accessible to authorized persons within the organization. For example, a local server within the organization. |
| (5) Digital signature means an electronic signature based upon cryptographic methods of originator authentication, computed by using a set of rules and a set of parameters such that the identity of the signer and the integrity of the data can be verified. | Digital Signature – An electronic signature that authenticates and verifies the signer’s identity and integrity of the records they sign. |
| (6) Electronic record means any combination of text, graphics, data, audio, pictorial, or other information representation in digital form that is created, modified, maintained, archived, retrieved, or distributed by a computer system. | Electronic Record – Digital information within a computer system. |
| (7) Electronic signature means a computer data compilation of any symbol or series of symbols executed, adopted, or authorized by an individual to be the legally binding equivalent of the individual’s handwritten signature. | Electronic Signature – A set of symbols that functions like a handwritten signature, but uses computer technology to sign the documents. |
| (8) Handwritten signature means the scripted name or legal mark of an individual handwritten by that individual and executed or adopted with the present intention to authenticate a writing in a permanent form. The act of signing with a writing or marking instrument such as a pen or stylus is preserved. The scripted name or legal mark, while conventionally applied to paper, may also be applied to other devices that capture the name or mark. | Handwritten Signature – Authentic and unique scripted name or legal mark created by an individual in writing. |
| (9) Open system means an environment in which system access is not controlled by persons who are responsible for the content of electronic records that are on the system. | Open System – Persons who control the records of the system, but do not control access to it. Meaning, the organization that generates the record doesn’t necessarily store it on their own and may be subject to outsourced data center/licensed software. That way, the records get accessible to only the authorized persons more widely through proper encryption. For example, outsourced remote cloud data solution. |
Subpart B – Electronic Records
11.10 – Controls for Closed System
| Regulation | Interpretation |
|---|---|
| Persons who use closed systems to create, modify, maintain, or transmit electronic records shall employ procedures and controls designed to ensure the authenticity, integrity, and, when appropriate, the confidentiality of electronic records, and to ensure that the signer cannot readily repudiate the signed record as not genuine. Such procedures and controls shall include the following: | Organizations using e-records in a closed system must meet the following characteristics: → Authenticity → Integrity → Confidentiality (when appropriate) → Unquestionable (impossible to dispute its authenticity) To achieve this, procedures and controls must contain the following information: |
| (a) Validation of systems to ensure accuracy, reliability, consistent intended performance, and the ability to discern invalid or altered records. | Computer System Validation: Conduct Computer System Validation (CSV) to ensure that the data processed in it is unquestionable. |
| (b) The ability to generate accurate and complete copies of records in both human readable and electronic form suitable for inspection, review, and copying by the agency. Persons should contact the agency if there are any questions regarding the ability of the agency to perform such review and copying of the electronic records. | Record Processing: An organization must be able to present e-records in both human- and computer-readable forms to the agency for inspection and copying. |
| (c) Protection of records to enable their accurate and ready retrieval throughout the records retention period. | Record Protection: Before the lifecycle ends, records must be readily available in their original form. |
| (d) Limiting system access to authorized individuals. | System Access Rights: Use a password protected system to ensure individuals only get to see what they are authorized for. |
| (e) Use of secure, computer-generated, time-stamped audit trails to independently record the date and time of operator entries and actions that create, modify, or delete electronic records. Record changes shall not obscure previously recorded information. Such audit trail documentation shall be retained for a period at least as long as that required for the subject electronic records and shall be available for agency review and copying. | Audit Trails: Each system that records GxP data must have audit trail functionality for all those actions (creation, modification, deletion) in local date and time without altering the earlier data. Additionally, it should: → Process the data in human readable form whenever agency demands. → Store the data as long as the record itself is stored. |
| (f) Use of operational system checks to enforce permitted sequencing of steps and events, as appropriate. | Sequencing: Prevent your electronic systems from doing activities in the wrong sequence. e.g., modifying the record after it has been digitally signed. In short, workflow of the digital systems is as per the GxPs. |
| (g) Use of authority checks to ensure that only authorized individuals can use the system, electronically sign a record, access the operation or computer system input or output device, alter a record, or perform the operation at hand. | Authority Checks: In line with the point (d) of this section, users that perform any activity in the system must be authorized to do so. |
| (h) Use of device (e.g., terminal) checks to determine, as appropriate, the validity of the source of data input or operational instruction. | Source Verification: A source from where the data is being added into the regulated system must be validated. |
| (i) Determination that persons who develop, maintain, or use electronic record/electronic signature systems have the education, training, and experience to perform their assigned tasks. | Personnel Qualification: Exhibit, the persons using the system for their respective purposes are qualified to perform their tasks. |
| (j) The establishment of, and adherence to, written policies that hold individuals accountable and responsible for actions initiated under their electronic signatures, in order to deter record and signature falsification. | Responsibility and Accountability: To discourage data falsification, SOPs must document responsibility and accountability criteria for actions done using E-signs. In addition, companies to maintain documented evidence (signed list of persons) as the persons using the digital systems understand that their E-sign is equivalent to the handwritten sign. |
| (k) Use of appropriate controls over systems documentation including: (1) Adequate controls over the distribution of, access to, and use of documentation for system operation and maintenance. (2) Revision and change control procedures to maintain an audit trail that documents time-sequenced development and modification of systems documentation. | Document Control: → Electronic systems, also, are eligible for Qualification, SOPs, Validation, Change Control, Deviation, etc. → So, it is the organization’s duty to deploy the electronic systems under such different document verticals. → System documents may include operational and maintenance procedures, audit trails, etc. |
11.30 – Controls for Open System
| Regulation | Interpretation |
|---|---|
| Persons who use open systems to create, modify, maintain or transmit electronic records shall employ procedures and controls designed to ensure the authenticity, integrity and confidentiality of electronic records as appropriate from creation to receipt. Such procedures and controls shall include the aspects covered in Sec. 11.10 as appropriate, and additional measures such as document encryption and use of appropriate digital signature standards to ensure, as necessary under the circumstances, record authenticity, integrity and confidentiality. | Companies using Open Systems must ensure their procedures and controls are: → Authentic → Integral → Confidential How do we achieve this in Open Systems? All the controls mentioned in Sec. 11.10 + additional security measures like document encryption and digital signature standards. Digital signature standards include: Name, Meaning, Date and Time, Impossible to falsify, Unique, Permanent, and Secure. |
11.50 – Signature Manifestations
| Regulation | Interpretation |
|---|---|
| (a) Signed electronic records shall contain information associated with the signing that clearly indicates all of the following: (1) The printed name of the signer; (2) The date and time when the signature was executed; and (3) The meaning (such as review, approval, responsibility, or authorship) associated with the signature. | E-signatures are able to capture signer’s: → Name → Timestamp, and → Meaning |
| (b) The items identified in paragraphs (a)(1), (a)(2), and (a)(3) of this section shall be subject to the same controls as for electronic records and shall be included as part of any human readable form of the electronic record (such as electronic display or printout). | The particulars mentioned in the previous point are subject to Part 11 and hence to be in human-readable form. |
11.70 – Signatures/Record Linking
| Regulation | Interpretation |
|---|---|
| Electronic signatures and handwritten signatures executed to electronic records shall be linked to their respective electronic records to ensure that the signatures cannot be excised, copied, or otherwise transferred to falsify an electronic record by ordinary means. | If an electronic record is signed (ink or electronic), the signature must remain associated with the record permanently. No action can be taken to erase, copy, or transfer it. |
Subpart C – Electronic Signatures
11.100 – General Requirements
| Regulation | Interpretation |
|---|---|
| (a) Each electronic signature shall be unique to one individual and shall not be reused by, or reassigned to, anyone else. | The system must be able to prohibit the duplication of E-signatures. |
| (b) Before an organization establishes, assigns, certifies, or otherwise sanctions an individual’s electronic signature, or any element of such electronic signature, the organization shall verify the identity of the individual. | It is the company’s responsibility to verify the identity of a person before issuing an e-sign. |
| (c) Persons using electronic signatures shall, prior to or at the time of such use, certify to the agency that the electronic signatures in their system, used on or after August 20, 1997, are intended to be the legally binding equivalent of traditional handwritten signatures. (1) The certification shall be submitted in paper form and signed with a traditional handwritten signature, to the Office of Regional Operations (HFC-100), 5600 Fishers Lane, Rockville, MD 20857. (2) Persons using electronic signatures shall, upon agency request, provide additional certification or testimony that a specific electronic signature is the legally binding equivalent of the signer’s handwritten signature. | Companies who want to make equivalence of the E-signs with ink signs, must convey this to the FDA. (1) Write a paper letter signed with ink signatures and send it to FDA. (2) If the FDA needs additional proof in considering E-sign and Ink sign as equivalent, companies must provide it. |
11.200 – Electronic Signature Components and Controls
| Regulation | Interpretation |
|---|---|
| (a) Electronic signatures that are not based upon biometrics shall: | E-signatures that do not follow biometrics need to: |
| (1) Employ at least two distinct identification components such as an identification code and password. (i) When an individual executes a series of signings during a single, continuous period of controlled system access, the first signing shall be executed using all electronic signature components; subsequent signings shall be executed using at least one electronic signature component that is only executable by, and designed to be used only by, the individual. (ii) When an individual executes one or more signings not performed during a single, continuous period of controlled system access, each signing shall be executed using all of the electronic signature components. | (1) consider Username and Password: (i) For the 1st login, user must enter a username and password. To track further activities, only password-authentication is sufficient. (ii) If logged-out, the user can sign in but he/she must again enter the username and password for each such sign. |
| (2) Be used only by their genuine owners; and | (2) Ensure the E-sign is performed by the original person who owns that signature. |
| (3) Be administered and executed to ensure that attempted use of an individual’s electronic signature by anyone other than its genuine owner requires collaboration of two or more individuals. | (3) If the original person is absent, his supervisor and system admin can jointly E-sign on behalf of that person. |
| (b) Electronic signatures based upon biometrics shall be designed to ensure that they cannot be used by anyone other than their genuine owners. | Only true owners can perform their biometric E-signs. |
11.300 – Controls for Identification Codes/Passwords
| Regulation | Interpretation |
|---|---|
| Persons who use electronic signatures based upon use of identification codes in combination with passwords shall employ controls to ensure their security and integrity. Such controls shall include: | E-signs using identification codes (username and password) must ensure their security and integrity including the following controls: |
| (a) Maintaining the uniqueness of each combined identification code and password, such that no two individuals have the same combination of identification code and password. | The system must prohibit username/password duplication. |
| (b) Ensuring that identification code and password issuances are periodically checked, recalled, or revised (e.g., to cover such events as password aging). | For the username and password-based system, it must do the password → verification → recall → revision (renew expired passwords) at regular intervals to create a more secure environment. |
| (c) Following loss management procedures to electronically deauthorize lost, stolen, missing, or otherwise potentially compromised tokens, cards, and other devices that bear or generate identification code or password information, and to issue temporary or permanent replacements using suitable, rigorous controls. | Procedures must be in place for the electronic deauthorization of token/device that is → Lost → Stolen → Missing → Potentially compromised username and password. Additionally, stricter controls must be outlined to avoid recurrence. Meanwhile, issuance of a temporary or permanent replacement of such tokens/devices will be provided. |
| (d) Use of transaction safeguards to prevent unauthorized use of passwords and/or identification codes, and to detect and report in an immediate and urgent manner any attempts at their unauthorized use to the system security unit, and, as appropriate, to organizational management. | In case of unauthorized login attempts, the system must be able to detect and report it promptly to the responsible authority. |
| (e) Initial and periodic testing of devices, such as tokens or cards, that bear or generate identification code or password information to ensure that they function properly and have not been altered in an unauthorized manner. | The password carrying tokens must be cross-checked before issuance + periodically to ensure that no unauthorized functions can occur. |
That’s all about step-by-step interpretation.
21 CFR Part 11 Assessment & Compliance
In today’s pharmaceutical landscape, regulatory scrutiny has become more rigorous, making compliance an absolute necessity.
Many companies struggle with interpreting the guidelines correctly, executing the requirements effectively, and documenting their efforts thoroughly.
Let’s break down the practical steps to achieve compliance and ensure your systems are FDA audit-ready.
The Key to Compliance: Right Interpretation, Execution, and Documentation
To achieve compliance, follow these four essential principles:
1. Right Interpretations
- Understanding the scope and applicability of Part 11 is crucial. Not all records fall under its purview.
- Clarify whether the regulation applies to your specific system or process.
- Misinterpreting the requirements can lead to either over-complicating your systems or leaving critical gaps.
- Quick Example: A Quality Assurance Manager from a leading pharma company shared that their team initially thought Part 11 applied to all their electronic data. After a thorough gap assessment, they realized that many records didn’t need Part 11 compliance, saving significant resources.
2. Right Execution
- Implement robust validation procedures for electronic systems to ensure data accuracy and integrity.
- Incorporate technical controls like secure user authentication and automated audit trails.
- Regularly update systems with evolving regulations and emerging technologies.
- Quick Example:
A major pharmaceutical manufacturer faced an FDA Warning Letter due to inadequate electronic signature verification. Their oversight? Failing to include a two-factor authentication process. After revisiting their system architecture, they introduced multi-level access controls, which significantly reduced compliance risks.
3. Rigth Reasons
- Document the reasoning behind your compliance approach. This includes why a particular electronic system was chosen or why specific validation steps were performed.
- Use risk-based rationale to justify decisions, especially when deviations occur.
- Quick Example:
During an FDA audit, a company was questioned about their electronic record storage practices. Their success lay in demonstrating a scientific rationale that linked storage conditions to data integrity.
4. Clear Acceptance or Rejection
- Document the validation outcomes and deviations with precise language (with Acceptance or Rejection).
Controlled Documents to Assure Compliance
To systematize the process, create two essential controlled documents:
21 CFR Part 11 Assessment Checklist
- Purpose: Helps determine whether a specific system requires Part 11 compliance.
- Contents:
- System Name and Description
- Data Type and Criticality
- Applicability of Part 11 Requirements
- Risk Assessment
- Example: For a laboratory information management system (LIMS), the checklist assesses whether data storage and audit trails meet Part 11 requirements.
21 CFR Part 11 Compliance Checklist
- Purpose: Confirms whether the identified system meets the regulatory standards.
- Contents:
- User Authentication Methods
- Audit Trail Maintenance
- Electronic Signature Verification
- Periodic Review and Validation Protocols
Why Integrate These Checklists into QMS?
Embedding these checklists into your Quality Management System (QMS) helps achieve:
- Streamlined Audits: Makes it easy to trace decisions during FDA inspections.
- Consistency: Ensures that compliance procedures are followed across different departments.
- Documentation Integrity: Provides documented evidence of compliance for audit readiness.
Industry Challenges and How to Overcome Them
Misinterpretation of Guidelines:
- Solution: Conduct internal training sessions and involve compliance experts for correct interpretations.
Overlooking System Updates:
- Solution: Schedule periodic reviews and updates as part of your QMS to ensure continuous compliance.
Lack of Adequate Documentation:
- Solution: Implement Standard Operating Procedures (SOPs) for GxPs.
Frequently Asked Questions and Answers
What is 21 CFR Part 11 compliance?
Any regulated information generated, accessed, stored, modified, or transferred; 21 CFR Part 11 compliance is required. It means, you must satisfy the FDA’s regulations in terms of managing your electronic records and electronic signatures.
How to decide if we require compliance with 21 CFR Part 11?
The best way to decide is to:
1. Create an assessment form as a question-answer sheet.
2. Attach this form as an Annexure to the relevant SOP.
3. Put key questions in the form. Answers should be Yes or No.
4. If any of the answers is Yes, you need to comply with 21 CFR Part 11.
5. The recorded form is now documented evidence of your decision. So, maintain this record for inspections.
Can we Contract Out?
Yes. But after all, you are accountable for your compliance.
Shall we customize the Audit Trail Reports?
You may find pre-formatted custom audit reports built into the system. Each moment of access (log-in, log-out, lock-out, activities) should be recorded and reported in an easy-to-understand format. The FDA may view these records during inspection and that’ll help them quickly find what they are looking for. Keeping audit trails in raw format will waste your as well as auditor’s time.
Part 11 doesn’t talk about lock-out, log-out? Why?
Some best practices automation experts use:
1. Automatically log-out the user after 5-10 mins. of inactivity.
2. System locks out the user after 3 failed password attempts and if the user is continuously inactive for specific days.
3. Setting up password complexity. At least, 1 Capital, 1 Special Character, 8 Characters, 1 Number etc.
You see, the regulations don’t mention these. But they do communicate one thing. Consider additional controls to make your systems more secure. That’s why right interpretation is the key to compliance.
We do every activity on paper. Shall we comply with 21 CFR Part 11?
Simply No. However, there are instances when electronically generated reports like test reports, trends, etc. that are printed and then ink-signed. For such instances, you need to have a documented rationale for not considering 21 CFR Part 11. It also applies to the paper records being shared electronically, uploaded on QMS, e-mails, etc. So, you probably don’t know but with such things, you may unknowingly become eligible to comply with 21 CFR Part 11. That’s why you see above an Assessment checklist to find out whether you need to comply with the regulation.
How E-sign can be linked to a document?
Signs must be encrypted with a private key assigned to each signer. Such digital signatures are considered safe and secure. Following are the non-compliances.
1. Scanning the signature and putting it into the record.
2. Screenshot manipulation of the physical signature.
These are violations under section 11.70 which interprets as: If an electronic record is signed (ink or electronic), the signature must remain associated with the record. No action can be taken to erase, copy, or transfer it.
How to Validate Electronic Systems under 21 CFR Part 11?
For this, the FDA recommends reading the following
1) General Principles of Software Validation; Final Guidance for
Industry and FDA Staff (https://www.fda.gov/media/73141/download)
2) GAMP 5 Guide 2nd Edition (https://ispe.org/publications/guidance-documents/gamp-5-guide-2nd-edition)
Go through these resources to interpret further.
The Bottom Line
Compliance with 21 CFR Part 11 is not a one-time effort. It’s an ongoing commitment to maintaining data integrity and regulatory adherence. By focusing on accurate interpretations, methodical execution, robust rationale, and clear documentation, you can establish a compliance framework that stands up to regulatory scrutiny.
Integrating assessment and compliance checklists into your QMS not only eases the burden of FDA audits but also reinforces your commitment to quality and patient safety.
Want more insights into how to tackle FDA compliance challenges? Leave your thoughts or questions in the comments below!






Hi Saket,
Is there any specific requirements for password complexity in any guideline?
Hi Surendra,
I would recommend you reading Section 11.300(a) to (e) for what FDA wants as password complexity requirements. Once you know WHAT, HOW becomes another story. Ensure access to electronic records and using digital signatures is limited to only authorized individuals.
For example, Section 21 CFR 11.300(b) states that it is important to ensure that identification codes and passwords are periodically checked, recalled, or revised to maintain the security and integrity of electronic records and signatures.
Interpretation: To enhance security, one method is to mandate that employees update their passwords regularly, such as every 90 days. This practice lowers the chances of unauthorized individuals gaining access to electronic data through illegal means like hacking. Furthermore, protocols should be established to promptly deactivate or change identification codes and passwords when an employee departs from the organization or suspects a security breach.