Cleaning Validation: The Definitive Guide

Manufacturing multiple drugs in the same facility is an efficient way to run the business. Yet, it involves risk of cross-contamination.
To avoid this, previous product traces must be completely removed before changing over the product.
This is called Product-to-Product cleaning.
Apart from this, a dedicated product facility also requires effective residue removal from batch to batch to avoid the carry over of impurities.
This is called Batch-to-Batch cleaning.
Cleaning validation is a scientific technique that ensures:
- Previous product traces have been removed
- Contamination-free operations
- Safe batch-to-batch transitions
In recent years, cleaning validation guidelines have become as complex as process validation due to raising concerns over the quality of the cleaning.
This article discusses the what, why, when, and how of this technique and the broad classification of available cleaning mechanisms.
Table of Content
What is Cleaning Validation?
Establishing documented, scientific and risk-based evidence that provides a high degree of assurance that a typical cleaning method or procedure will consistently clean the equipment or a medical device in compliance with its predetermined specifications and quality attributes, taking the patient’s safety into consideration. In a nutshell, it is a technique of developing effective cleaning processes that’ll not harm the end-user.
This is the “essence” grasped from various regulatory guidelines.
Why Cleaning Validation Matters (Big Time)
Let’s get one thing straight:
Cleaning validation isn’t a major headache for single-use systems. You toss them out after use – no cross-contamination risk there.
But for reusable systems? Especially in India?
It’s a hot zone.
Regulatory heat is rising.
So are product complaints.
And guess what’s often to blame?
Poor cleaning.
So… Why Bother with Cleaning Validation?
When you manufacture a drug, residues stick around.
It could be:
- API traces
- Raw materials
- Excipients
- Even leftover cleaning agents
Now picture this:
You run the next batch. Or worse, switch products.
Boom… Cross-contamination.
And that’s exactly what you’re trying to prevent.
Cleaning Validation Is About One Thing:
Proving your cleaning process works.
Not assuming.
Not hoping.
Proving.
It’s about showing there’s no chance of product contamination.
That’s why regulators expect it.
And that’s why your QA team won’t greenlight anything without it.
But There’s More To It Than Just “Cleaning Well”
A smart cleaning validation program must plug into:
- Your Quality Management System (QMS)
- Your Quality Risk Management (QRM) framework
Why?
Because it helps you:
✅ Identify risks
✅ Understand their impact
✅ Build mitigation strategies
✅ Execute them
✅ And ultimately, protect product quality and patient safety
What is Contamination?
The product that is adulterated with the residue of the previous batch of the same product in particular equipment is called Contamination OR A new product that is adulterated with the residue of raw materials of the previous product is also called Contamination.
What is Cross-Contamination?
A new product that is adulterated with the left behind traces of the previous product in particular equipment is called Cross-Contamination.
What Makes Cleaning Validation a Win?
- Patient Safety: Backed by science and risk-based studies, your cleaning process ensures safety.
- No Contamination: Avoid product adulteration and guarantee a clean, contamination-free product.
- Regulatory Compliance: Meets all the necessary regulatory standards.
- Optimized Processes: Streamline product change-overs and cut unnecessary costs.
Now, let’s dive into…
- The types of cleaning processes and how they work
- How to build a cleaning validation program from scratch
- The expectations of regulatory bodies you need to know
When to Perform Cleaning Validation?
The following are the situations that triggers the requirement of a proper cleaning validation study.
- When establishing a fresh commercial process
- When reusing the existing facility for a different product every time
- Major changes in raw materials based on impact assessment
- Significant modifications in cleaning procedures
- Introduction of new equipment for the already established process
- Changes in cleaning agent
- Changes that might affect the CPPs and CQAs of already approved cleaning validation
Cleaning Classifications
Cleaning processes based on industrial practices can be differentiated into three ways and two types.
3 Ways of Cleaning
Cleaning can be performed in 3 different ways using cleaning agents such as purified water, WFI (Water for Injection), or chemical solvents.
- Cleaning In Place (CIP)
- CIP Skid to Clean the Equipment
- Automated CIP of the Equipment
- Cleaning Out of Place (COP)
- Washers like Tray Washers, Dish Washers, etc.
- Sub-systems of equipment hard to approach in CIP
- Manual Cleaning
- Components that are difficult to clean during COP and hence cleaned using tools such as Cleaning Brushes, Scrubbers, etc.
Not a thumb rule but in general:
- Equipment is preferred for automated cleaning in place i.e., CIP.
- Components and sub-systems not being feasible for CIP are preferred for COP.
- Critical components or locations of the equipment that are hard to reach during CIP and COP are preferred for Manual Cleaning.
2 Types of Cleaning
As per the common understanding among pharma professionals, there are two types of cleaning.
- Batch To Batch
- Cleaning of the process equipment in between two batches for ongoing manufacturing campaign of the same product.
- Product to Product
- Cleaning of the process equipment in between the two different products i.e. after finishing the previous product campaign and before initiating a new product campaign.
Different Cleaning Mechanisms
Residues from equipment can be removed either physically or chemically. It is chosen by consistency and performance-based selection criteria.
- Physical
- Mechanical or Manual
- Emulsification
- Dissolution
- Chemical
- Solubilization Using Solvent
- Chemicals
- Detergents
Mechanical Cleaning
This cleaning involves the mechanical application of scrubbers, brushes, wipes, etc. for removing residue or previous product traces.
Let us see one by one.
Brushing or Scrubbing
- Detergent solutions are prepared with a slightly alkaline pH in hot water, typically NLT 50°C.
- The loose components are disassembled from the equipment and dipped in detergent solutions allowing sufficient soaking time.
- A brush, scrubber, or scrapper as suitable is applied on the surface and rinsed under hot or cold water for sufficient time.
| Advantages | Disadvantages |
|---|---|
| Simple and flexible | Inconsistency due to manual action |
| Requires exhaustive training | Involves additional man-power |
Water Spray
This involves a high-pressure water spray that easily disintegrates the residue from loose and dirty components of the equipment.
Moreover, when water alone is insufficient to do this job, a suitable surfactant is considered for further scrubbing or brushing solutions.
Wiping
Lint-free cloths are commonly used to clean visible equipment surfaces with wiping actions defining:
- Wiping directions, patterns, and number of times to repeat the wiping
- Solvent and cleaning agent concentrations
- Quantity of cleaning solution to apply to the wipes
Emulsification
Hydrophobic residue suspended in an aqueous cleaning solution is called Emulsion. This method is typically considered while dealing with insoluble liquid residues and is rarely applicable in pharmaceutical industries.
However, firms that follow detergent-based washing cycles may still find this method effective. Though considered a physical cleaning mechanism, it may also fall under Detergents.
Surfactants are introduced in the equipment to form an emulsion. When agitated, break down the residue into small droplets and either float or sink based on the density.
Practically speaking, enough variations are possible in this method due to the uncertainty of the holding time of the residue.
Dissolution
In this mechanism, a residue is dissolved with a suitable solvent. The common solvent readily available is water which is aqueous in nature.
Non-aqueous solvents are also preferred in specific cases. However, water is non-toxic, cheap, environmentally friendly, does not contribute to chemical degradation, and is easy to remove. Hence water is the primary choice.
But choosing either of them depends on the drug residue solubility characteristics.
Chemicals
This mechanism involves the application of chemical reactions for cleaning purposes and generally includes oxidation and hydrolysis.
Oxidation
Strong oxidizing agents are used to break carbon-carbon bonds resulting in smaller molecules and increasing the water solubility of the residue.
Hydrolysis mentioned below too has the same impact but on a more specific level. Whereas oxidation is a universal term and hence puts challenges in choosing a specific analytical method to detect unoxidized residue.
Examples of oxidizing agents include Peracetic Acid (CH3CO3H), Hydrogen Peroxide (H2O2), and Sodium Hypochlorite (NaClO), etc.
Hydrolysis
This method improves the solubility of solvents resulting in hydrolyzed residue with lower molecular weights.
For aqueous solutions, hydrolysis is carried out at elevated temperatures using acids or alkalis. The rate of hydrolysis depends on the nature and quantity of residue and temperature.
It is important to consider the most appropriate analytical methods to test the efficiency of the cleaning process independently.
Detergents
Wetting
This method lowers the surface tension of residue with the help of surfactant addition in water. Wetting results in two actions.
- Wet residue reduces its surface tension and improves the rate of dissolution
- Wet equipment surface promotes residue disintegration
Dispersion
Dispersion is the same as that of emulsification except for the use of solid particles.
Solid particles are wetted and broken down using anionic surfactants and vigorous agitation to form a suspension pool. More importantly, this method is practiced in Oral Solid Dosages (OSD) such as power blending and tablet manufacturing.
Emulsification
Refer to the Emulsification Above.
Solubilization
In some cases, acids or alkalis are used for proper dissolution, yet they require excess removal times to bring them down to neutral pH ranges.
For example, bringing down the pH should be checked at the “drain point” rather than “in-place” to ensure complete residue removal from the equipment during cleaning.
These methods can either be used in combination or sequence depending upon the feasibility and nature of residue. It is highly advised to properly conduct a residual study to determine an effective cleaning mechanism.
List of Cleaning Agents
Organic Solvents
Generally, organic solvents are used in the bulk manufacturing of Active Pharmaceutical Ingredients (APIs).
When cleaned with water, reactors with higher volumes may not dissolve the bulk residue stuck to the surface of reactors.
An organic solvent used in manufacturing the same drug usually dissolves the API and hence the most suitable choice for cleaning purposes.
However, addressing cost optimization and solvent recovery are crucial aspects while designing the cleaning processes. Examples of commonly used solvents are Methanol, Toluene, Acetone, and Ethyl Acetate.
Aqueous Agents
Water is used as a stand-alone cleaning agent or in combination with the following classifications.
| Commodity Chemicals | Specialty Cleaners |
|---|---|
| Inorganic Chemicals – Ortho-Phosphoric Acid (H3PO4) – Nitric Acid (HNO3) | Surfactants (Based on the charge on the polar end) – Nonionic (no charge) – Anionic (+ve) – Cationic (-ve) – Amphoteric (+/- based on pH) |
| Organic Chemicals – Acetic Acid (CH3COOH) – Sodium Hydroxide (NaOH) | Other Cleaners like Chelants, Dispersants, etc. |
What we’ve understood so far was related to cleaning mechanisms and their commercially available options. You can visit this link for a list of cleaning agents. For now, that’s all about the cleaning philosophy.
Cleaning Validation Program
This is the most critical part of your manufacturing activity.
Since if the cleaning is inadequate, it may compromise the patient’s safety. Hence, cleaning processes should be designed and developed considering this as the 1st worst-case.
Identifying the core risks associated with the cleaning philosophy is the next important challenge. Risk identification requires systematic study instead of experience-based predictions.
Following are the most common CPPs and CQAs for a typical cleaning process.
Critical Process Parameters (CPPs) for Cleaning
- Temperature
- Pressure
- Contact time
- Concentration of the cleaning agent
- Final rinse conductivity
- Surface Roughness (More roughness means more difficult to clean)
- Flow rate
- Proper mixing RPMs
- Dirty Hold Time for Equipment
- Clean Hold Time for Equipment
- Number of rinses (could be a Key Process Parameter (KPP))
Critical Quality Attributes (CQAs) For Cleaning
- Product residue
- Cleaning agent residue
- Microbial residue
- Time for cleaning
Pre-Requisites to Begin Cleaning Validation
Before commencing cleaning validation, the following pre-requisites should be met:
- Cleaning Validation Strategy and Protocols are approved and ready
- Equipment used for most of the products should be identified
- SOP for Equipment Cleaning has been established (draft)
- Sampling and Analytical Methods are validated
- A list of CPPs and CQAs is available
- Drug toxicity data is available
- Product contact surface area is calculated
- Training is given to the personnel involved in cleaning validation activity
- Drug characteristics are evaluated for cleaning difficulty
Cleaning Validation Process Flowchart
Below is a flow diagram showing the overall strategic activities in cleaning validation in the pharmaceutical industry.
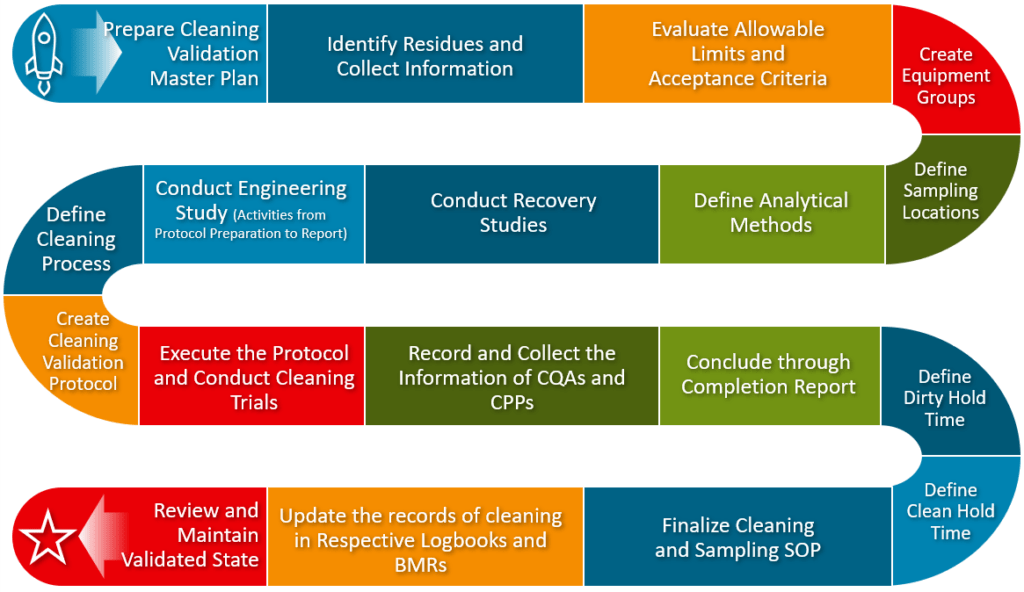
You may cascade the items as per the suitability of your cleaning process.
A risk-based approach would help identify the risks associated with:
- Concerned residue
- Priority of selecting sampling locations
- Determining and justifying product/equipment groups
- Defining acceptance criteria
- A sequence of protocol execution
- Product change-over SOPs
Cleaning Validation Protocol and Report
Instead of following any protocol template, it is important to understand its key technical aspects. A protocol should be prepared explaining technical cleaning validation activities that contain:
- Scope and Objective
- Cleaning SOP Draft
- Possible ways of contamination
- Sampling plan and rationale including prioritized sampling locations
- Worst-case conditions
- Selection of analytical methods and their validation
- Equipment Dirty and Clean Hold Time Study
- Acceptance criteria
- Technically sound deviation management strategy
- Data monitoring Annexures (if Applicable)
Successful execution of the protocol would be incomplete without a report. A report should be prepared to summarize the key achievements of the cleaning validation study, including a clear statement of acceptance/rejection.
Selecting a Proper Way of Cleaning
As the manual cleaning and COP are already discussed, let us only see CIP.
Clean In Place (CIP)
To meet the regulatory expectations and a promising cleaning, this is the most suitable way of cleaning the equipment. Equipment is cleaned at its existing location with a suitable cleaning agent.
It is recommended to develop and implement an automated cleaning sequence to better control the potential variations. This helps to achieve cleaning consistency and adapt to an identical cleaning pattern.
For Process Vessels and Integrated systems, a static or dynamic spray ball is preferred to ensure complete coverage of 360° including dead spaces near nozzles (one of the major worst-case locations).
A Riboflavin test is conducted to conclude that the spray ball is effectively reaching remote spaces of equipment at a defined pressure and flow rate. The Riboflavin solution acts as a speck of dirt and can be seen under Ultra-Violet (UV) light.
A typical CIP cycle generally includes the following rinses:
- Pre-rinse with PW or WFI
- Alkali and Acid Wash (if product demands)
- Final Rinse with PW or WFI
A proper air flushing mechanism should also be incorporated to remove stagnant water from the system avoiding chances of microbial growth.
Selection of Analytical Methods and Their Validation
Two fundamental types of analytical methods exist.
- Specific
- Non-Specific
The selection of these methods requires a science- and risk-based approach. Inappropriate evaluation and selecting any of these methods may invite regulatory objections.
For example, the FDA states that companies should determine the specificity of their analytical method. Most professionals misunderstand that the FDA expects to use only specific methods. On the contrary, this also means the selection of analytical methods requires proper evaluation.
However, the EU-GMP mentioned, “the analytical method must be specific for the target residue“. This may be interpreted in two ways.
- Only specific methods are acceptable OR
- Any of the methods must be specific to that particular target residue even with the non-specific method.
However, the second one sounds more logical and that’s the reason European drugmakers generally practice it.
Needless to say, there is much depth to it. Hence, a correct assessment of the method selection is very important to establish a scientifically meaningful cleaning process.
For easy understanding:
| Specific Methods | Non-Specific Methods |
|---|---|
| Gives us the exact quantification of the target residue | Related to the target residue but doesn’t provide its direct measurement or quantification |
| Recommended during cleaning validation | Recommended after cleaning validation OR also complement specific methods during cleaning validation |
| Can detect interference in substances other than residue like cleaning agents | Itself can falsify the measurement of the target residue because of the presence of other substances |
| Examples: HPLC, RP-HPLC with UV, Ion Chromatography | Examples: pH, Conductivity, Visual Detection, and TOC |
To bridge the gap, many pharmaceutical manufacturers follow the general practice i.e. a combination of both methods. Specific methods for primary cleaning validation while using non-specific methods for subsequent cleaning verification.
These methods can also be selected based on the stage of manufacturing. Initial stages such as bulk drugs or intermediates, excluding toxic APIs, may only use non-specific methods that may suffice the requirement. Whereas formulation and downstream processing may require both.
But the correct way is to conduct scientific studies and then choose the analytical method/s that accurately detects the compound of interest.
Analytical Method Validation
Upon successful selection of the analytical method, it is important to validate it for the intended use. All non-pharmacopeial analytical methods require validation.
However, if you want to use any of them even when a pharmacopeial method exists, you’ve to provide a rationale for doing so.
Just to clarify, methods described in the monograph of different pharmacopeias are called Pharmacopeial methods, and selecting those methods is evidence-based and need not require a formal method validation.
Anyway, the following characteristics should be covered during the analytical method validation.
- Specificity OR Selectivity
- Ability to use analytical methods to accurately measure analytes and interferences such as cleaning agents.
- Selectivity is checked with blank samples (without an analyte) examined in the anticipated time range of the peak that contains the analyte.
- Accuracy (% Recovery)
- Degree of agreement of the test results produced by the analytical method to the true value. Accuracy is generally established for a complete specified range of the procedure.
- A known concentration of analyte standard spikes the sample matrix and measures the accuracy using the specified analytical method.
- Precision
- The degree of agreement between the individual test results for the repeatedly applied analytical procedure to multiple samplings.
- It can be calculated by different methods like Statistical, Horwitz equation, etc.
- Limit of Detection
- The lowest concentration at which the instrument detects an analyte but does not necessarily quantify it. The noise to signal ratio should be 1:3.
- Limit of Quantitation
- The lowest concentration at which the instrument both detects and quantifies an analyte. The noise to signal ratio should be 1:10.
- Linearity
- The capability of the analytical method to obtain the outcome is directly proportional to the concentration of the analyte within a given range.
- 5 concentrations at a minimum are preferred from 50% to 150% across the working range and are injected with a mobile phase to produce a linear relationship.
- Range
- It is the concentration range and an interval between the upper and lower limit shown using precision, linearity, and accuracy.
- Stability of Solution
- The time duration of the sample for which it can be stored before the final analysis after extraction.
- Ruggedness
- It is a measure to determine the robustness and reliability of the analytical method to deliver linear, accurate, and precise results in all anticipated conditions.
Sampling Methods for Cleaning Validation
According to the FDA guidelines, there are 3 types of sampling. Out of that, 2 are commonly followed for cleaning validation.
Direct Surface Sampling (Swab Sampling)
Direct surface sampling (swab sampling) is the most preferred sampling method for hard-to-reach but reasonably accessible areas of the equipment.
A sterile swab made of cotton is attached to a compatible stick just like earbuds.
- The challenge is to use the solvent along with the swab comfortably without interfering with the analytical test. Swabs may contain certain adhesives that can alter the results.
- These swabs are stored and dipped in a buffer like a phosphate solution or as appropriate to soak the cotton. During sampling, swabs are removed and then applied gently on the equipment surface in one verticle and one horizontal direction without rubbing to and fro.
- Once done for at least 5 to 6 sampling locations, they are put back into the buffer solution and sent to the Quality Unit for evaluating and establishing an acceptable residue content per given surface area as CFU/cm2 or as appropriate.

Though this method is more specific in terms of hard-to-reach areas, it has one major disadvantage i.e. the small surface area for a given sample.
Rinse Sampling
Unlike swab sampling, rinse sampling has the advantage of covering a large surface area of the equipment in a particular instance, including systems that are hard to disassemble frequently.
- The required amount of cleaning solvent with the help of a suitable spray ball preferably with 360° coverage used to rinse the equipment.
- Rinsed samples are then collected from sample points located near drain lines for physical and microbiological inspection.
One of the major disadvantages of rinse samples is when rinsed, the residue may incompletely solubilize in the rinse solvent like WFI or PW and remain clogged to the equipment surface. In this case, just checking downstream water for compendial requirements is illogical and hence unacceptable.
Instead, the system should be in place to identify the direct measurement of the residue in the rinse sample such as Infrared sensors or visual inspection, etc.
Indirect Testing or Monitoring Method
Although it is a sampling method or more specifically a monitoring method, it is also an indirect measurement that does not specifically provide us with exact quantification of the residue.
Hence not acceptable as a stand-alone sampling method during cleaning validation. Instead, it should either be used as complementary to the actual sampling method or should be routinely verified after the cleaning validation program.
It is more specific in cases like bulk drug manufacturing where sampling can be most easily performed through rinsing. The best examples are pH, conductivity, and TOC (Total Organic Carbon) measurements.
However, in special cases where other methods fail to detect any presence, this method can be scientifically justified.
Points to Remember while Selecting Cleaning Validation Sampling Locations
1. Picking What’s Easy Over What’s Risky
Let’s face it: Professionals commonly pick the most accessible spots.
But regulators aren’t interested in what’s easy to clean.
They want to know you’ve checked where the gunk actually hides.
Dead legs. Welds. Crevices. Hard-to-clean spots.
That’s where you should be swabbing.
2. No Risk Assessment = No Direction
Jumping into sampling without a risk-based approach?
That’s like flying blind.
Evaluate equipment geometry, material, past residue issues and the works.
Sampling should be guided by data, not guesswork.
3. Changing Sampling Points Like Socks
Sampling points aren’t something to switch up every cycle.
Inconsistent locations = inconsistent results.
Pick your worst-case spots and stick with them.
4. Assuming “Looks Clean” Means “Is Clean”
Visual inspection is nice.
But it won’t catch 2 ppm of potent residue.
Use validated analytical methods or risk failing audits.
5. Skipping the Why Behind the Where
Ask most teams why they chose a sampling spot.
You’ll get blank stares.
Document your rationale.
It shows you’ve thought it through and gives auditors confidence.
6. Neglecting the Equipment’s Unique Personality
A centrifuge is not a granulator.
Yet many use the same sampling plan across all equipment.
Bad move.
Design matters. Tailor your sampling plan to the machine’s quirks.
7. Tossing Out Gold: Historical Data
Your past cleaning validation holds hidden gems.
Don’t ignore it.
Dig in.
Find trends, problem areas, and use it to refine your current strategy.
Defining Equipment’s Dirty Hold Time and Clean Hold Time
Everything in the world comes with an expiry and cleaning is not an exception to this. Therefore, the timeframe for the cleaning validity and dirty conditions becomes crucial. But what do they mean?
- The equipment’s idle time between the end of the last batch and the start of the cleaning process is called Dirty Hold Time.
- The equipment’s idle time between the end of the cleaning process and the start of manufacturing is called Clean Hold Time.
Dirty Hold Time
Dirty hold times are the most crucial aspect of a cleaning validation program as they directly impact the efficiency of the cleaning process.
When equipment is left uncleaned for longer durations, the residue attached to the surface may become rigid and dry over a period of time, ultimately challenging the cleaning process.
Establishing dirty hold times for a particular production process depends on the nature of the product, associated processing materials, and cycle times.
The general practice is to conduct 3 consecutive runs of cleaning procedure considering maximum dirty hold time as per the requirement (in most cases around 72 hrs.).
These runs should demonstrate the cleaning procedure effective in removing the residue at the considered maximum dirty hold times. Obviously, through bioburden testing.
According to one of the FDA’s 483 observations, cleaning validation and dirty hold times should be established for dedicated as well as non-dedicated equipment. This should also include hard-to-clean equipment to obtain overall confidence in cleaning validation.
Clean Hold Time
Just like dirty hold times, the FDA also expects to define clean hold times during the cleaning validation program.
Clean Hold time study generally includes a sampling of clean equipment at a regular time interval of around 6 to 8 hrs. till the equipment completes 24 hrs.
After 24 hrs., the sampling is done once per day. Sampling is performed immediately after cleaning and thereafter at specified intervals.
It is better to have a data recording sheet that captures the necessary information when the samples are sent to the QC lab for bioburden testing (microbiological proliferation).
Data Recording Sheet Example:
| Cleaning Hold Time | Nature of Analysis | Acceptance Criteria |
|---|---|---|
| 0 hrs. | Microbial: CFU/Swab Chemical: PPM/Swab | Depends on the application for example, Microbial: NMT 10 CFU/Swab Chemical: NMT 10PPM/Swab |
| 6 hrs. | —“— | —“— |
| 12 hrs. | —“— | —“— |
| 18 hrs. | —“— | —“— |
| 24 hrs. (Day 1) | —“— | —“— |
| Day 2 | —“— | —“— |
| Till the required time | —“— | —“— |
Worst-Case Conditions in Cleaning Validation Program
Conditions that may critically impact cleaning validation efforts or even cause a failure should be identified with the most specific challenges.
Following are the challenges considered for establishing the worst-case conditions.
- Drugs with the lowest solubility in their cleaning agent
- Swab locations that are difficult to clean
- Lower therapeutic doses for effective cleaning measurement
- Equipment catering for the largest number of products
- Drugs with higher toxicity
Establishing Cleaning Limits and Requirements
Poorly defined acceptance criteria can undermine your entire cleaning validation efforts.
Two key concepts you need to know? NOEL and MACO.
No Observed Effect Level (NOEL) tells the drug quantity that has no observable effect on human health when provided with a 50% Lethal Dose.
Maximum Allowable Carryover (MACO) tells you mathematically how much of your previous product will carry over to the next product.
a) Calculating NOEL
NOEL can be calculated as:
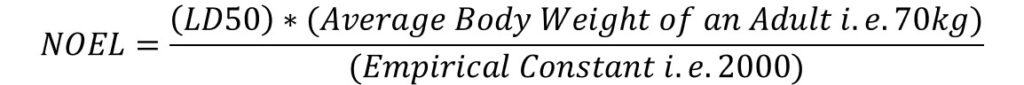
Where LD50 – Lethal Dose is at 50% reduction in mg or kg and NOEL is generally measured in “mg”.
However, this approach of risk identification is pointless to carry forward. According to one of the European Medicines Agency (EMA) Question and Answer documents, the use of LD50 to determine Health-Based Exposure Limits (HBEL) for drug products is an inadequate point of departure.
b.1) MACO Based on Therapeutic Daily Dose and Safety Criteria
Based on the above calculated NOEL values, the MACO values can be calculated as:

As per this criterion, no more than 0.1% normal therapeutic dose of the previous product shall appear in the maximum daily dose of the next product.
Here, the min. batch size is considered for the next product.
b.2) MACO Based on 10 PPM Criteria
As per this, no more than 10 ppm of the previous product shall appear in the next product.
MACO can be calculated as:

b.3) Calculating MACO Using Toxicity Data
This approach is generally considered during the early stages of drug manufacturing such as Intermediates or APIs. Additionally, this technique is used to calculate MACO for cases that don’t have information regarding the therapeutic dose.

Strict control of cleaning agent residues is a critical part of pharmaceutical manufacturing. Since these agents come into direct contact with production equipment, any leftover residues can lead to cross-contamination risks.
Let us break down the key methods for setting residue limits to ensure product purity and regulatory compliance.
Regulatory Framework and Health-Based Exposure Limits (HBEL)
The European Medicines Agency (EMA) emphasizes the importance of setting Health-Based Exposure Limits (HBELs) to ensure patient safety. In its April 2018 update, the EMA introduced the term “medicinal,” creating a clear distinction between medicinal products and non-medicinal substances like cleaning agents.
This change suggests that while HBELs may not be directly mandated for non-medicinal substances, pharmaceutical manufacturers are still expected to assess and establish appropriate limits. Understanding these guidelines is crucial for ensuring compliance and maintaining safe manufacturing practices.
By carefully interpreting EMA regulations, companies can navigate these requirements effectively while upholding high-quality standards in pharmaceutical production.
What is HBEL?
HBEL stands for Health-Based Exposure Limit.
It’s the maximum amount of a residual substance (like an API or cleaning agent) that can remain on equipment or in a shared facility without causing harm.
Think of it as a safety threshold that ensures cross-contamination doesn’t put anyone at risk.
How to Calculate HBEL?
HBEL is calculated using the Acceptable Daily Exposure (ADE) formula:
HBEL (µg/day) = (NOAEL)*(BW) / (UF)*(F)
Where:
- NOAEL = No Observed Adverse Effect Level (from toxicology studies)
- BW = Body Weight (standard is 50 kg for humans)
- UF = Uncertainty Factors (accounting for data gaps)
- F = Adjustment Factors (like bioavailability)
Practical Example:
Say a drug has a NOAEL of 10 mg/kg/day from animal studies.
Using a safety factor of 100 and a human body weight of 50 kg, the HBEL would be: HBEL = 10×50/100 = 5mg/day
This means a person can safely be exposed to 5 mg of the substance per day without adverse effects.
Why HBEL?
Before HBEL, companies used outdated 1/1000th dose methods that overestimated risk.
Now, HBEL provides a science-driven, risk-based approach that ensures safety without unnecessary restrictions.
Grouping Strategies: : Why They Don’t Work Anymore
Back in the early 2000s, grouping strategies were the go-to method for simplifying cleaning validation. The idea was simple: group similar equipment or products, validate one representative, and call it a day. It was all about saving time and boosting productivity.
But here’s the problem: it didn’t work.
Fast forward to today, and the FDA and other regulatory bodies are not buying it. During audits, experts realized that these grouping practices actually compromised patient safety. Instead of making cleaning easier, they introduced more cross-contamination risks.
Why Grouping Strategies Fail
- Unfit Justifications: Auditors found that the reasoning behind grouping wasn’t scientific enough.
- Blaming Personnel: Instead of reassessing the grouping logic, companies often blamed cleaning failures on human error. FDA didn’t accept that excuse.
- Safety Concerns: Bottom line, grouping strategies were putting patients at risk.
The result?
More observations. More compliance issues. More finger-pointing.
Many healthcare manufacturers are ditching outdated grouping practices and focusing on science-based, risk-driven cleaning processes. Patient safety matters way more than just saving a few hours.
Traditional Grouping Strategy: What Was the Plan?
The idea was to validate one representative operation and use that as proof that similar operations were also clean. This would minimize cleaning validation efforts – a tempting shortcut.
But here’s the kicker:
The FDA never had a specific policy endorsing grouping strategies. They always wanted a solid, scientific rationale. And that’s where most manufacturers failed.
Indian Manufacturers: Time to Catch Up
It’s time for Indian healthcare manufacturers to move on from outdated practices. Focus on building robust, risk-based cleaning processes that actually ensure patient safety.
Because in the end, saving time is great – but not at the cost of patient safety.
The Strategy
The traditional grouping strategy was:
Validating one representative operation to demonstrate its effectiveness on all similar types of operations is called a grouping strategy.
Successful cleaning validation of that representative operation would establish that all the associated grouped operations are also validated.
This strategy minimizes cleaning validation efforts i.e. cleaning identical equipment and products. The FDA does not have a specific policy for considering grouping strategies, but the FDA recommends a scientific rationale (a big exercise to worry about) for such grouping strategies.
Grouping mostly could be of two types.
- Product Grouping
- Equipment Grouping
Product Grouping
Product grouping can be performed based on 3 different criteria. Instead of going theoretical here, let’s look at this. Please zoom in or open the image in a new tab for better reading.
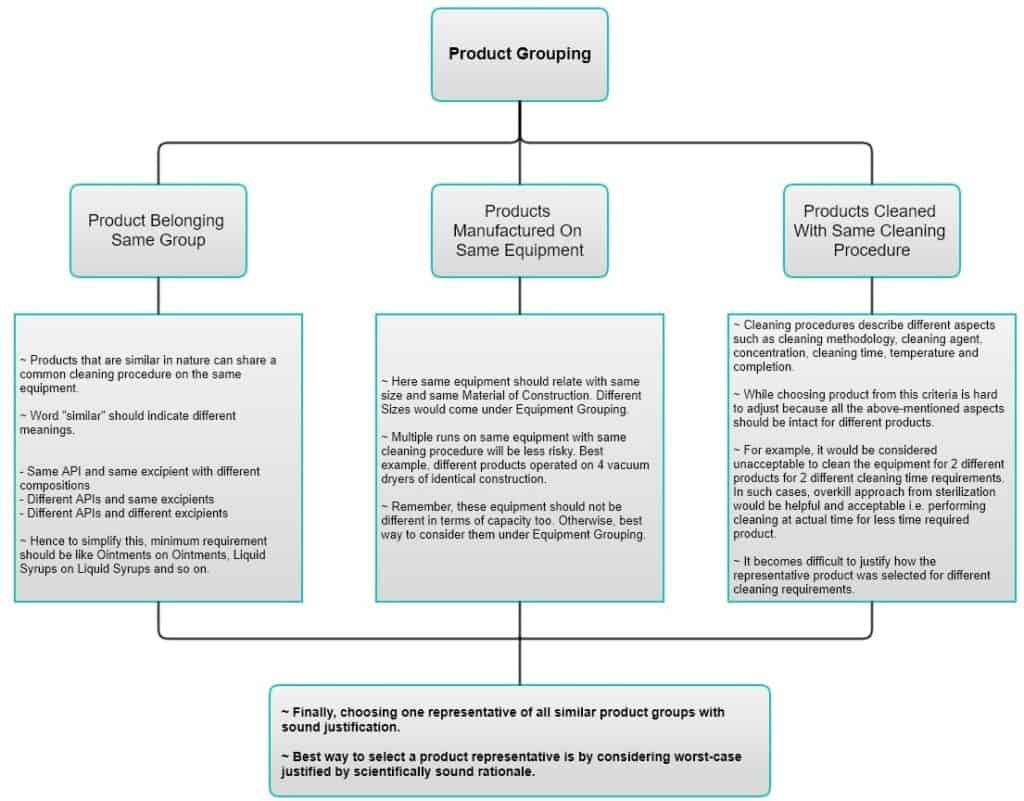
Equipment Grouping
Unlike Product Grouping, this technique is used to perform cleaning on one representative piece of equipment.
The first and foremost condition of equipment grouping is that all similar equipment follow the same cleaning procedure.
Additionally, it is not acceptable to group different equipment with the same cleaning mechanism and application e.g., Vacuum Tray Dryer and Roto-Cone Vacuum Dryer. Though the ultimate purpose and cleaning philosophy is the same, the structure and dimensions of the two pieces of equipment are completely different.
Equipment must be similarly designed to fall under equipment grouping. And that similarity has to be established.
A simple example of equipment grouping is process vessels of different working capacities. Here, “similar design” should indicate different aspects of equipment design such as MOC, Geometry, sub-components, and so on. Simply put, one cannot group the equipment of Stainless Steel (SS) and Glass Lined Reactors (GLR).
However, a major challenge in the example above is selecting a representative vessel based on worst-case conditions.
How would you justify a small capacity vessel as harder to clean than a large-capacity vessel?
In such cases, the combination of equipment should be selected for cleaning validation. That is, performing cleaning on the equipment group with one smaller volume vessel and one larger volume vessel. Otherwise, one can select two pieces of equipment separately for multiple cleaning runs individually.
Both scenarios are assumed to establish an overall degree of assurance for the effectiveness of the cleaning procedure.
Practicality of Using Grouping Strategies
Grouping strategies, here, are discussed only for understanding purposes. The development of cleaning processes requires a risk- and science-based agenda.
When identifying risks (not superficial but fundamental), systematic risk mapping is very important instead of gathering experienced anticipations. Firms have to conduct studies in assessing and identifying the potential risks of the facility being unclean. Then they set their action plan accordingly.
Cleaning Validation Regulations and Guidelines
Let’s be real.
Most pharma companies don’t fail audits because they don’t clean.
They fail because they can’t prove their cleaning works.
Here’s what inspectors often say:
“Your cleaning validation is inadequate.”
“You failed to prevent cross-contamination.”
“Your equipment cleaning program is not scientifically justified.”
Ouch.
But here’s the kicker…
It’s rarely about visible dirt.
It’s about invisible gaps in your cleaning strategy.
What Regulators REALLY Want?
They’re not expecting magic.
They’re expecting logic.
✅ Risk-based cleaning approach
✅ Clear rationale for cleaning agents and methods
✅ Scientifically justified acceptance limits
✅ Worst-case product assessment
✅ Solid lifecycle documentation
That’s it.
No spoon-feeding.
No second chances.
Why Most Cleaning Programs Fail?
Let’s break it down.
#1: No risk assessment
#2: No rationale for “why this method”
#3: Arbitrary limits with zero science behind them
#4: Documentation that doesn’t speak for itself
And guess what?
No agency gives you product-specific residue limits.
But they WILL question your logic behind the numbers.
Here’s What Auditors Look For
They open your protocol and expect answers. Fast.
- Why this cleaning method?
- Why this limit?
- Where’s the worst-case logic?
- What happens if equipment stays idle?
- How do you handle visual vs. analytical methods?
No answers = No mercy.
Use These Guidelines (Or Get Burned)
Here’s your cheat sheet:
- FDA – Cleaning Validation (2021 draft)
- EMA – Annex 15
- PIC/S PI 006-3 – Cleaning Validation
- WHO TRS 1019 – Annex 4
- ICH Q9(R1) – Risk Management
- ISPE Baseline Guide Vol. 7
- Health Canada GUI-0028
Know them. Apply them.
And build a program that can defend itself.
What Audit Observations Reveal (That Guidance Doesn’t Say Out Loud)
Let’s decode what regulators really want…
Based on what they actually write in warning letters.
1. Document Your Validation Philosophy
“The company’s overall policy and approach to validation should be documented.”
– EU GMP Part II, Section 12.1
This includes:
- Cleaning procedures
- Analytical methods
- In-process checks
- Roles and responsibilities
If your approach isn’t clearly mapped, expect questions.
2. Poor Equipment Design = Guranteed 483
“CP-2 packaging line was modified… the line cover isn’t removed during line clearance.”
– FDA Warning Letter 06-NWJ-14
Translation?
If your equipment makes cleaning hard, you’re already failing.
3. Don’t Just Swab. PROVE Your Method Works
“Sampling methods must be challenged to show recovery.”
– EU GMP Annex 15, Section 4.10.3
Negative swab results mean NOTHING…
Unless you can prove recovery is consistent and robust.
4. Use Sensitive Analytical Methods
“Detection limit should be sufficient to detect the acceptable level.”
– ICH Q7A
Your method should be able to see what you claim to control.
5. Document Test Specificity
“Integrated peaks near standard retention time must be identified.”
– FDA 483 Observation
If your peaks overlap and you can’t tell one from another, you’ve got a specificity problem.
6. Sloppy Cleaning Validation = Sloppy Audit Outcome
“Protocol didn’t define cleaning procedure. Swab validation missing. Rinse samples not analyzed.”
– FDA 483 Observation
That’s not just one issue.
It’s a laundry list of red flags.
7. Design for Cleanability
“Valves, filters, and pipework should facilitate cleaning and sterilization.”
– EU GMP Annex 2
“Avoid dead legs. Minimize residue accumulation.”
– PIC/S Annex 9
“Washing equipment should NOT be a source of contamination.”
– PIC/S Chapter 3
If your system has dead legs, pooling spots, or blind ends…
You’re practically inviting contamination.
8. CIP ≠ Clean by Default
“Identify critical areas – especially in semi or fully automatic CIP systems.”
– EU GMP Annex 15, Section 4.6.1
Automated doesn’t mean foolproof.
Spot-check the hardest-to-clean parts and prove they’re clean.
9. Hose Handling = High Risk
“Flexible hoses lying on the floor increase contamination risk.”
– FDA Guide to Inspections of Oral Solutions and Suspensions, 1994
Don’t just worry about cleaning hoses.
Worry about how they’re handled, stored, and connected.
Coiled hoses = trapped moisture = microbial growth.
Cleaning Validation Compliance: What You Must Do
If you’re serious about getting (and staying) compliant, here’s your blueprint.
1. Document Everything
Robust SOPs.
Clear cleaning protocols.
Calibration records.
If it’s not documented, it didn’t happen.
2. Train Like You Mean It
One-time training? Not enough.
Run regular sessions focused on:
- Equipment handling
- Contamination control
- Cleaning validation fundamentals
- PPE usage for hazardous drugs
Training should build competence, not just tick a box.
3. Validate Every Step
Cleaning isn’t about “looks clean.”
It’s about data that proves it’s clean.
So:
- Validate your cleaning procedures
- Validate your sampling methods
- Validate your analytical techniques
- Validate performance over time
No shortcuts.
4. Control Cross-Contamination Like a Pro
Strengthen your containment strategy:
- Facility and equipment design
- Proper hose management
- Cleaning agents and frequencies
- Hazardous drug handling protocols
And yes: PPE training is non-negotiable.
5. Own Your Equipment Lifecycle
Stick to a strict calibration and maintenance schedule.
Design for easy cleaning (zero dead legs).
Document everything: calibration, maintenance, inspection.
6. Monitor and Improve Continuously
Implement in-process checks.
Use environmental monitoring to catch issues early.
Audit your cleaning systems (internally) before regulators do.
Meaning?
– Start with documentation.
– Back it up with training.
– Prove it through validation.
– And keep it alive with monitoring.
Agencies vs. Associations: Know the Difference
Here’s a pro tip:
Regulatory agencies = legal authority.
Think: FDA, EMA, CDSCO.
They enforce compliance.
Industry associations = expert consensus.
Think: ISPE, PDA.
They provide guidance, not law, but often influence how regulations are interpreted.
You need to know both.
Conclusion
Cleaning validation isn’t just about scrubbing tanks.
It’s about proving (with data) that your process works.
Every time. No shortcuts.
✅ Set clear, measurable cleaning goals.
If it can’t be defined, it can’t be validated.
✅ Choose and validate your analytical method wisely.
Sensitivity and specificity aren’t optional, they’re your foundation.
✅ Consolidate your findings.
Wrap it up with a report that summarizes:
- Pass/Fail outcomes
- Closed deviations
- Lessons learned
✅ Don’t just meet expectations. Investigate for improvement.
Use the data to tighten your process, not just tick the box.
✅ Make it risk-based and science-driven.
Combine development logic with real-world execution.
That’s what builds a resilient cleaning strategy.
Over to you.
What’s your biggest challenge in cleaning validation?
Ever had an audit surprise you with something unexpected?
Drop your thoughts below. Let’s talk shop.






Hi Saket,
I have doubt about the manual cleaning runs during validation. In case of manual cleaning runs, the outcome of critical cleaning parameters (cleaning time, water quantity etc.) could be differ from batch to batch results and its obvious due to operator variability (Different operators) for example :
Cleaning Step: X Run -1 Run-2 Run-3
Cleaning Time 15 min 35 min 17 min
Purified Water quantity 30 lit 45 lit 33 lit
Please note no optimization/development/trial data was available.
In this case, what should we do:
Either we should consider max. parameters (Max time/water quantity ) or need to recommend for further validation or both?
Could we use the first run outcome as guidance for execution of next two run ?
I hope you understood my concern and help me to find the solution.
Hi Surendra, without having any optimization/development/trial data I doubt how you will be able to validate your cleaning process. There must be some assumptions and science-backed info that would have helped you arrive at the current cleaning process. And regarding cleaning variations due to manual process, you can recommend max parameters for further validation. Either way, as the process still manual, you will get repeated variations. Bandwidth of those variations can be assumed and defined. Otherwise it will go under deviation. 1st run outcome can be used as a guidance. But as I said, variations are bound to happen. Better you define their acceptance in your cleaning validation program.
Rinse sampling with purified water/WFI for the molecules which are insoluble doesn’t make any sense to verify the contamination. Use of some selective solvents based on method validation is a more useful technique for rinse sampling, however I have not found any such recommendation or restriction in any guideline. Could you help me to answer this.
Adding to this verification of solvent used for sampling shall be the part of study.
Hi Surendra, I can understand your confusion about solvent selection and method validation. PW or WFI are just examples provided above, please read again. Solvent selection if you see, requires scientific approach to ensure the product residue getting dissolved into it while rinsing. Surprised to know how you weren’t able to find this in any guideline for that sake FDA’s Validation of Cleaning Processes. Some products do get dissolved with PW/WFI. It depends how you study it and conclude.
Thank you, this is a very useful overview of the major aspects of Cleaning Validation.
I would like to make a few comments on the CQA / CPP section though:
– equipment design elements cannot be CQAs or CPPs, as for instance drainability does not say anything about the cleaning process but about the design of the equipment to be cleaned. It is equipment qualification that proves that the system is suitable to validate a cleaning process on
– at some points, the CQAs and CPPs are mixed up, cleaning agent concentration is a CPP, not a CQA in my opinion, because we control the concentration to meet the CQA of process residue removal. The final rinse conductivity tells us that the cleaning agent is removed from the system, so that is a CPP. The number of rinses that is required to achieve the threshold value of this CPP, could be a KPP (Key Process Parameter) at best.
Happy to discuss this off-line
Thanks, Marco for the value addition. You are almost correct. The suggestions and the comments are now implemented and article is revised.
People like you make this platform more useful to other professionals. Keep doing the great work!
This is a very useful article thank you! For dedicated equipment with no degragants, is the expectation to perform a Health based calculation for product residues limit?
Hello, may I use your material as base for instructional purposes?
I usually perform calculation of MAC using PDE for liquid sterile drugs. An inspector made a recommendation saying that we have to compare the results with minimum dose and choose the lower.
Why do I have to use a less scientific method when there is nothing more scientific than PDE?
I’m sure using PDE, that the toxicological aspect are considered. Why do I have to put limits lower if my calculation has a scientific and toxicological basis?
At this point I can choose the LOQ of the method if I have to obtain the lowest!
Dear Sir. Thank you so much for the well writen document on Cleaning Validation.
Hello Sir,
If our products are only excipients and it does not have specificity for analysis,
please suggest what to do.
Is it necessary to perform recovery test?
Hi Ravindra, as far as you only produce excipients and specificity for analysis isn’t applicable, similarly recovery test isn’t applicable too.
It is really great work Mr. Saket,
can we consider the stopper bowl and chute in equipment grouping while perform the dirty equipment. as per guidelines product contact parts will be consider for equipment grouping but stopper bowl and chute are not direct product contact parts.
Hi Venkat, your two comments (this one and previous one) looks contradictory. Please confirm you question properly.
it is great work, during dirty equipment hold, we consider the product contact parts like manifold, filling pumps and nozzels, can we consider the stopper bowl and chute in equipment grouping while perform the dirty hold? My concern is the stopper bowl and chute are in direct product contact parts.
This article was really helpful. Could you specify how to clean an equipment after defined clean hold time runs out (it is not used for 20 days but the clean hold time is 10 days). Should we do the whole cleaning procedure again or can we just take a sample for microbiology and clean it with alcohol on the surface it the test is good (no bacteria found). Thanks in advance
Thanks, Mikasa for the appreciation. Regarding your query, it clearly looks a case of deviation where you need to log the deviation, investigate the failure to meet dirty hold time requirement, assess the impact, analyze, propose CAPA and close the same with sound rationale. Because, the whole purpose of the dirty hold time study would have been compromized.
Great article on cleaning validation. What are your thoughts regarding cleaning validation requirements on reusing a previously dedicated line for a new product. The line would now only be dedicated to the new product. What are the expectations in this situation. Both products have established cleaning validations for dedicated equipment. There would be that initial time where equipment was previously used for and dedicated for product A, but now dedicated to only new product B.
Thanks, Mary for the appreciation. Regarding your question: As you are talking about product changeover for the existing facility, you will need to perform entire cleaning validation activity. Ensure that the product A traces are removed entirely eliminating chance of cross-contamination. Suggest you to follow this article for detailed break-down of activities.
Hi Mr. Saket, thanks for sharing this clear and detailed article, glad to read this. I have question about the maintaining cleaning validation state, should we conduct routine validation or such periodic cleaning verification to ensure the validation state is well maintained? Could you please share any guideline talk about this part? Thanks a lot.
Thanks for your appreciation. Regarding your question, FDA talks about Continued Process Verification that also applies to your cleaning procedures.
There are various guidelines that explains how to ensure the consistency of your already validated cleaning process. You can search for:
– FDA, Guide to Inspection of Validation of Cleaning Processes
– EC, EudraLex Volume 4, EU Guidelines for Good Manufacturing Practice for Medicinal Products for Human and Veterinary Use
– PIC/S, PIC/S Validation-Master Plan, IQ, OQ, Non-sterile Process Validation, Cleaning Validation (PI 006-3) (September 2007)
Let me know how it goes.
Wonderful information Saket. Could you pls clarify if cleaning verification needs to be performed after every cleaning to ensure lproper cleaning, specially in case of multiproduct equipment?
Thankyou
Vaishali
Thanks, Vaishali! Regarding your query, verification is a different thing.
You must verify if the cleaning you performed between batch to batch of the same product. Of course, with the reduced efforts than the cleaning validation since your cleaning process is already established as an effective one.
To give you simple example, you perform cleaning validation for batch to batch and steps roughly should be:
1. Defining cleaning goals
2. Strategy and draft SOP
3. Protocol
4. Execution (Actually monitoring the cleaning parameters)
5. Verifying the cleaning termination conditions
6. Summarizing the findings
7. Conclusion via report and making SOP effective.
Whereas, once this is done and while you are doing routine cleaning batch to batch.
1. Following the cleaning SOP
2. Verifying the cleaning termination conditions are met.
Hope this explains your doubt clearly.
It’s really very useful information to better understand the process. Can you please make one on equipment qualification as well?
Thanks, Alex for your kind words. I already have one published for Process Validation that includes the qualification part.
Here are the links for quick access:
https://pharmagxp.com/quality-management/process-validation/
https://pharmagxp.com/quality-management/qualification-and-validation/
Wow… Great job… Excellent Material… Thanks very much!
Glad you found this excellent, Rama! Best of luck.
Dear sir,
I have read about the MACO calculation in your article and i have some quesy about it as per below.
1. For any equipment such as S
Tank, rinse and swab both sample are taken. So what will be the chronology of sample ?
Swab for micro then chemical swab then rinse sample ???
2. If we have more than one cleaning agent ,then does the MACO for active residue will be different or all product shall be considered under same train ??
3. In maco calculation, LARGEST DAILY DOSE OF NEXT PRODUCT is considered.these dose is therpetutic dose of next product for API or unit dose of next product.
Such as one tablet of next product’s weight is 500 mg and api content per tablet is 250 mg..then Larget dose in calculation will be 250 mg or 500 mg ??
4. Once the maco calculation is done and validation is completed . Now during review it was found that MACO value is incorrect and after recalculation ,MACO value found below LQL value of analytical method.. then what shoud we do ??
5. Does 100ug per square cm for swab is limit for visual inspection ? Or it should be eatablishe by protocol based study !! Does any article or guideline support this ?
If calculated MACO value is higher than visual inspection limit (100ug per sq.cm swab ) , then based on visual inspection ,cleaning should be verified visuaaly and equipment will be released ..is it true ? Or still validation need to be performed ??
6. What is the microbial limit for Swab and rinse ??does any pharmacopoeial or other guideline limit available ?
Hope you understand and give your valuable response.
It’s very useful, well explained with observations received from FDA. Thank You for such an article on cleaning validation…
Glad you liked it, Mahesh!
Clear and interesting. Can you please let me know which guidelines clearly states the cleaning level type?
Thanks, Heba for admiring the content. However, I don’t think if such types of levels are mentioned in any guidelines. Rather, regulations like FDA do mention the types of sampling, methods of analysis, and establishment of limits.
The content was great. I enjoyed it and thank you
Glad you find it helpful.
The article written by you gives the great information collectively. It is very informative and helpful to everyone.
I really enjoyed reading this article.
Thanks, Vilas. Glad you find the content informative!😊
I enjoyed a lot while reading the article. Good theory. Thank you for sharing such data with us…
Pleasure all mine, Srinivasa 😊
Exceptional work, thoroughly enjoyed reading it.
Appreciate your words Luis 🙂
Really an excellent effort Mr. Saket. I can say you are the library of your profession! 😊 I relished the reading.
Super satisfied with your feedback Rakesh 😊 Best wishes!
I enjoyed the write-up on Cleaning Validation. More power to your elbow.
Glad you found the content useful 🙂