FDA 483s and Warning Letters: Mastering Compliance for Overseas Pharma Sites (OUS)

76% of FDA Warning Letters in the last 3 years went to overseas pharma sites.
That’s not a trend.
That’s a target.
If you’re operating an OUS site, one FDA 483 can trigger a supply chain disaster, import alerts, or even a total shutdown.
But here’s the good news:
You can decode the patterns, understand what FDA expects, and build systems that not only pass inspections but also elevate your global compliance reputation.
In this report, you’ll see:
- The repeat violations killing compliance
- Why most “inspection prep” fails
- A real strategy to build lasting audit readiness
If you manufacture outside the US, this is your wake-up call.
Here’s a graph showing the rise in the Warning Letters to overseas (OUS) pharma sites, segmented by region (India, China, EU, Brazil).
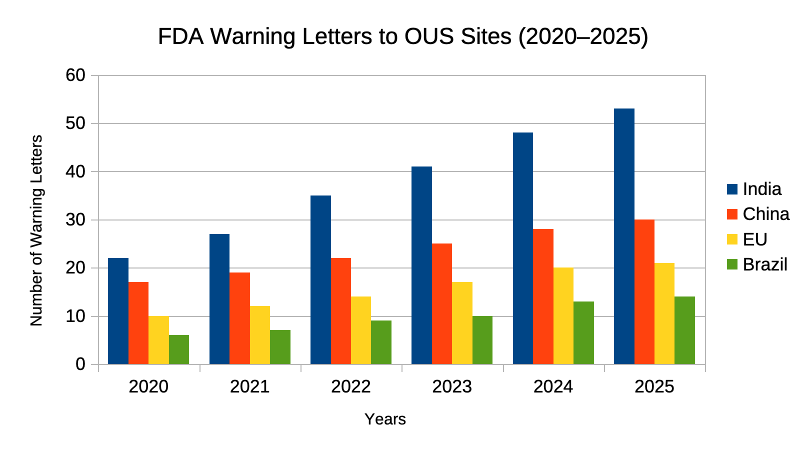
Source: Approximation from FDA.gov Warning Letters & Third-Party regulatory trackers.
Table of Content
Officially: FDA Now Launching Unannounced OUS Inspections
In a major policy shift (May 6, 2025), the FDA announced it will begin conducting unannounced inspections at overseas manufacturing sites producing drugs, devices, and essential medicines. Historically, foreign sites received advance notice unlike their U.S. counterparts but now this parity aims to expose concealed deficiencies and level the global playing field.
“For too long, foreign companies have enjoyed a double standard—given advanced notice before facility inspections, while American manufacturers are held to rigorous standards with no such warning. That ends today. This is a key step for the FDA as part of a broader strategy to get foreign inspections back on track,” said FDA Commissioner Martin A. Makary, M.D, M.P.H.
“The FDA’s rigorous, science-based global inspections of manufacturing facilities ensure that the food and drug products that enter the U.S. marketplace, and the homes of American consumers, are safe, trusted, and accessible,” said FDA Assistant Commissioner for Inspections and Investigations Michael Rogers. “These inspections provide real-time evidence and insights that are essential for making fact-based regulatory decisions to protect public health.”
Big Shift: No More Heads-Up for Overseas Pharma Sites
Starting May 2025, the agency will ramp up unannounced inspections at foreign manufacturing sites, especially those supplying critical drugs and medical devices to the U.S.
Why?
To close the visibility gap between domestic and overseas audits. Too many OUS facilities clean up their act just before a pre-scheduled inspection.
Key takeaway?
If your site isn’t inspection-ready 24/7, you’re at risk.
This is no bluff. The FDA is deploying more foreign office staff, expanding language capabilities and collaborating with global regulators to enforce this new model.
👉 Official FDA press release here
That’s why mastering compliance for FDA 483s and Warning Letters is your frontline defense.
Chapter 1: The Anatomy of a FDA 483 – What It Really Means
A FDA 483 is a form of errors and a reflection of systemic gaps in your site’s GMP compliance.
Quick Breakdown:
| Field | What it Means |
|---|---|
| Form FDA 483 | List of observations from FDA during inspection |
| Warning Letter | Issued post FDA 483 if response is inadequate or violations are severe |
| Import Alert | Site banned from shipping to US unless lifted |
Key Insight: 8 out of 10 warning letters to OUS sites are related to data integrity and inadequate investigation systems.
The Real Problem: It’s Not Just What the FDA Finds
Form FDA 483s list what’s wrong.
But that’s just the surface.
The deeper issue: Systemic failures that keep showing up, again and again.
We’re talking:
- Broken data integrity practices
- Ineffective Quality Units
- Poor contamination control
These aren’t one-off misses.
They’re signs of a quality system that’s not built to last.
And the FDA sees it.
That’s why they’re tightening the grip on overseas facilities.
Want to stay off their radar?
Fix the system. Not just the symptoms.
Data Integrity: The FDA’s Main Red Flag
When the issued a Form FDA 483 or Warning Letter, one theme pops up over and over:
Data Integrity.
We’re talking about:
- Missing or incomplete records
- Lab data that doesn’t match
- Weak access controls
- No audit trails
- Files sitting on external hard drives
Sound familiar?
Take the recent Warning Letter to Center Instrumental Analysis, China Pharmaceutical University.
The FDA found:
- Lab records stored outside the system on external drives
- No restrictions on who could access or change NMR data
- A complete breakdown of control over critical information
That’s a direct hit to ALCOA+ principles, the global gold standard for trustworthy data.
If data isn’t:
- Original
- Enduring
- Attributable
- Accurate
- Available
Then it’s not just a technical glitch. It’s a regulatory landmine.
Here’s the kicker:
Many pharma sites are buying high-end lab instruments…
But skipping the part where they secure, govern, and validate the data those systems produce.
That’s a big mistake.
Because if the FDA can’t trust your data. They can’t trust your product.
And if they can’t trust your product, they won’t trust your site.
Simple as that.
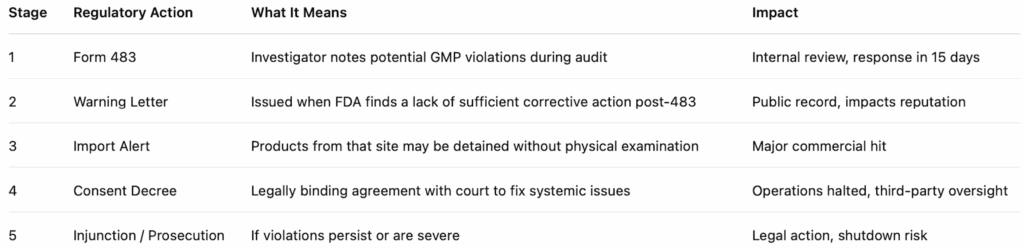
Here’s a simple ladder of regulatory actions:
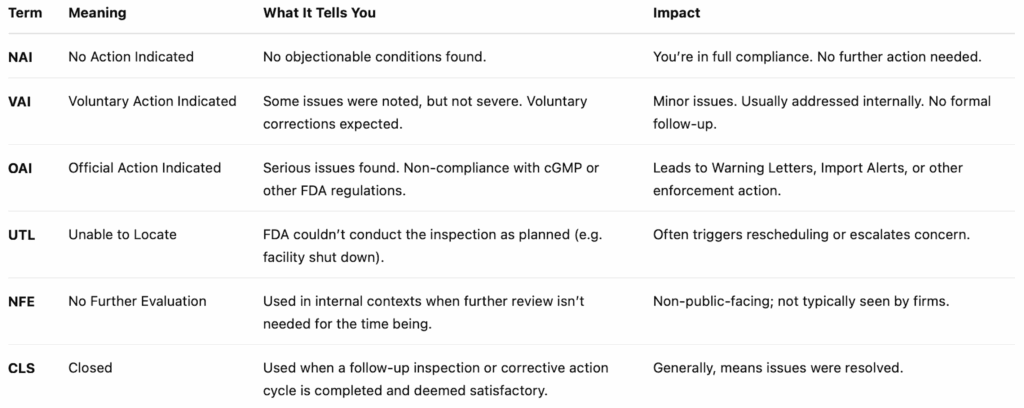
And here are the FDA audit classification terms that signal the outcome of an inspection. They’re used internally and also show up in public databases like the FDA’s inspection classification database:
Cleaning Failures: The Silent Compliance Killer
Cleaning validation is a CGMP fundamental.
But here’s the ugly truth: Most firms are still getting it wrong.
Recent FDA inspections uncovered jaw-dropping issues:
- Thick residues on equipment untouched for days
- Swab samples with APIs 800x the acceptable limit
- Chromatograms riddled with unidentified peaks
Let that sink in. These’re not only dirty machines but also risk to compliance.
So what’s going wrong? Apart from bad execution, it’s bad validation design.
Here’s what the FDA keeps calling out:
- Incomplete lab records
- Sampling procedures that miss the real risk zones
- Investigations that go nowhere
- Visual inspections treated like gold standards
The result: Contamination hides in plain sight.
Your cleaning may “look” good. But without solid validation, it’s no sense.
And if your lab can’t even identify what’s showing up in chromatograms, you’ve already lost the quality game.
Here’s the fix:
- Validate smarter, account for hard-to-clean zones
- Use science, not guesswork, in sampling design
- Demand analytical methods that detect, not deflect
- Document everything like your license depends on it (because it does)
Cleaning failures are definitely residue issues.
But they’re trust issues: with regulators, with patients, and with your brand.
Your move: Is your cleaning validation really science- and risk-based. Or just a wrap?
Here are the common FDA 483 observations & their systemic implications for OUS Pharma:
| Observation Category | Specific Examples/Details (from FDA findings) | Systemic Implication |
|---|---|---|
| Data Integrity | Incomplete lab records, unauthorized data changes, data on external drives, lack of controls on computerized systems | Digital blind spot, erosion of trust in data reliability, flawed data governance, compromised patient safety |
| Quality Unit Deficiencies | Lack of QU authority/resources, inadequate oversight of investigations, insufficient approval of quality decisions | Organizational disempowerment, breakdown of quality oversight, reactive rather than proactive quality management |
| Cleaning & Contamination Control | Thick residues in equipment, unknown peaks in chromatograms, swab samples exceeding limits, inadequate cleaning sampling, incomplete cleaning validation records | Invisible contamination, flawed validation design, analytical gaps, direct threat of cross-contamination and microbial contamination |
| Inadequate Investigations | Failures to investigate deviations, recurring issues, lack of root cause analysis | Reactive compliance, breakdown of Corrective and Preventive Action (CAPA) systems, inability to prevent recurrence |
| Equipment Qualification | Unqualified equipment, lack of authorization controls on computerized systems, poor maintenance | Compromised product quality and patient safety, regulatory non-compliance, unreliable manufacturing processes |
Chapter 2: Why Overseas Sites Are at Higher Risk
Here’s the cold truth: Overseas pharma sites face a steeper hill when it comes to FDA inspections.
Not because they’re less capable. But because their systems aren’t calibrated to FDA’s exacting lens.
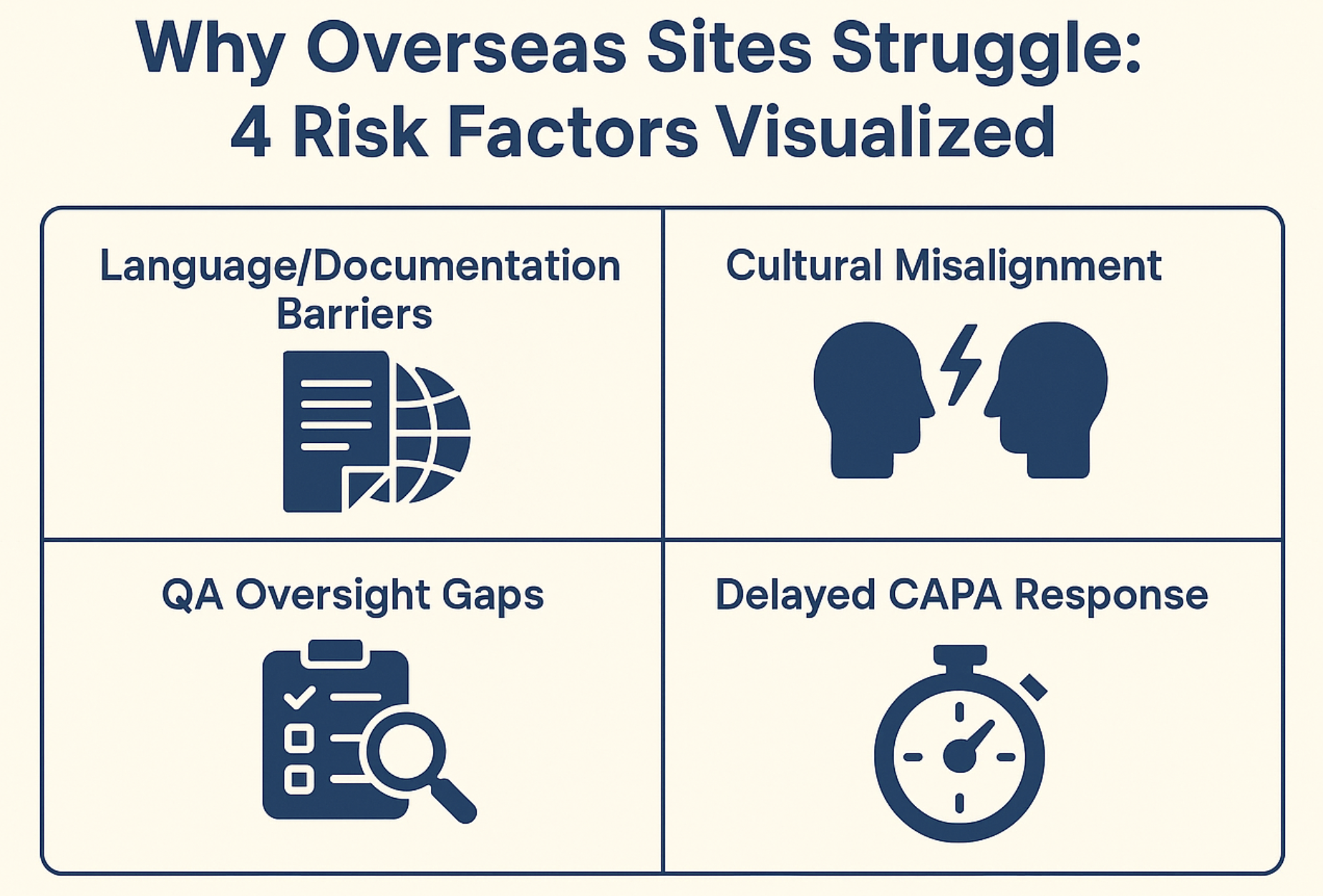
1. Language Barriers = Documentation Gaps
FDA inspectors don’t have time to “interpret.”
If SOPs, test records, or batch sheets are mistranslated, inconsistent, or vague, you’ve already lost credibility.
- A missing word.
- An ambiguous instruction.
- A poor translation of “immediate action” as “future improvement”.
That’s how FDA 483s start.
2. Cultural Misalignment with FDA Expectations
In many regions, hierarchical culture leads to “Do as told”.
But the FDA expects critical thinking, challenge culture, and real root cause analysis.
So when investigators ask, “Why didn’t QA stop the batch?”
And the answer is, “Because Production said it’s fine”.
That’s a red flag.
FDA expects independent QA.
Not QA that follows the org chart.
3. Weak Internal QA Oversight
At some OUS sites, QA is treated like a “document control” department.
They just push the paperwork and stay out of the way.
That won’t fly.
The FDA wants QA that can:
- Halt batches
- Drive investigations
- Own cleaning validation
- Verify data integrity controls
Weak QA equals weak control.
And weak control gets you Warning Letters.
4. Sluggish CAPA Response Timelines
In the U.S., CAPA responses move fast.
OUS? Not always.
It’s common to see:
- 30+ day delays in root cause analysis
- Action plans with vague timelines
- No effectiveness checks ever
The FDA doesn’t care if “it takes time to align teams”.
They expect timely and measurable fixes. Not bureaucratic inertia.
Real-World Example
An Indian API site received 5 observations.
- 3 were for missing and manipulated lab data
- 2 were for shallow OOS investigations
Issue was their response was late and vague.
Result:
- Warning Letter
- Import Alert
- Millions lost in contracts
Bottom Line:
Being outside the U.S. is not an excuse.
It’s a responsibility multiplier.
Want to stay FDA-ready?
- Write every SOP like an FDA investigator will read it tomorrow
- Empower QA to lead, not follow
- Build a CAPA system with speed, substance, and follow-through
- Train teams to think in “FDA speak” and not just local norms
Global operations demand global-level compliance. No shortcuts.
Chapter 3: The Top 5 Observation Themes (Based on Last 5 Years)
The numbers don’t lie.
We analyzed hundreds of FDA 483s issued to Overseas Pharma Sites (OUS) over the last 5 years.
Here’s what came up again and again:
| Sr. # | Observation Theme | % Occurrence in OUS FDA 483s |
|---|---|---|
| 1 | Data Integrity | 76% |
| 2 | Inadequate Investigations (OOS/OOT) | 61% |
| 3 | Poor Equipment Cleaning Validation | 54% |
| 4 | Batch Records Not Reviewed | 47% |
| 5 | Vendor Qualification Gaps | 39% |
| 6 | Sterility Assurance | 39% |
| 7 | Formalized CAPA Programs | 25% |
| 8 | Supplier Controls | 15% |
| 9 | Laboratory Controls | 12% |
| 10 | Quality Unit Authority | 9% |
What’s the Pattern?
These aren’t clerical errors. They’re not “one-off oversights”.
They’re system-level failures.
Every single one points to gaps in how quality is designed, enforced, and lived inside the facility.
Let’s break them down:
1. Data Integrity (76%)
Still the #1 issue, by far.
Why?
Because most firms still:
- Store critical data on unsecured drives
- Lack audit trails on instruments
- Have weak access control
- Can’t prove entries are ALCOA+
Translation? FDA can’t trust your data and ultimately the product.
2. Inadequate OOS/OOT Investigations (61%)
This is classic.
Batch fails a spec.
Investigation starts.
And instead of root cause analysis, they write:
“Retested. Passed. No further action.”
That’s not an investigation.
That’s a cover-up.
FDA expects:
- Clear Phase I and Phase II segregation
- Hypothesis-based approach
- Full documentation of impact on other batches
- Science-first thinking
If you’re just “testing into compliance,” expect a FDA 483.
3. Poor Equipment Cleaning Validation (54%)
Visual inspection does not mean VALIDATION.
Especially when FDA finds:
- Swabs with residues 800x above the limit
- “Uncleaned parts” in manufacturing tanks
- Chromatograms with unidentified peaks
- Missing hold-time studies or dirty rinse data
Your cleaning validation is not only about GMP but it’s your defence against cross-contamination.
Get it wrong and you’re risking patient safety and product approval.
4. Batch Records Not Reviewed (47%)
This one’s simple.
But deadly.
- Missing QA signatures
- No reconciliation of yields
- Data added after-the-fact
- Entries out of sequence
- BMRs archived without review
Your batch record tells the FDA how that product was made.
If it’s incomplete, unaudited, or altered – You’re signaling you don’t control your process.
5. Vendor Qualification Gaps (39%)
Your product is only as strong as your weakest supplier.
FDA’s finding:
- No vendor risk ranking
- Outdated audit reports
- No written agreements on quality responsibilities
- No qualification of critical material suppliers
In one case, an API site used an unqualified vendor for solvents.
The solvent failed purity tests. And the drug was recalled in three countries.
Think external vendors don’t matter? Think again.
Bottom Line:
These 5 patterns point to a simple truth:
FDA doesn’t cite documentation. They cite dysfunction.
You can’t “format” your way out of a FDA 483.
You need a living, breathing Quality System that:
- Catches failures before FDA does
- Investigates deeply
- Documents everything with traceable intent
- Fixes your system + SOPs
- Reviews batch records like product release depends on it (because it does)
- Audits and qualifies vendors like they’re part of your facility
Chapter 4: How to Read a FDA 483 Like an Investigator
Most firms read a FDA 483 and start patching SOPs.
That’s the wrong move.
To actually fix the issue, you need to read a FDA 483 like the FDA wrote it.
Here’s the blueprint.
1. Decode the Language
Every word in a FDA 483 is intentional.
FDA inspectors don’t write casually but legally.
Look for these red flags:
- “Your firm failed to…” → Critical breakdown. Systemic. Repeated or ignored in past inspections.
- “Procedures were not followed…” → Breakdown in execution. SOP exists but isn’t working.
- “Your SOP did not adequately…” → Procedural gap. Your system exists but lacks robustness.
Pro tip:
Anytime you see “failed to ensure,” expect scrutiny in future inspections.
That phrase often shows up again in a Warning Letter.
2. Map Each Observation to a System
You don’t fix FDA 483s by editing documents.
You fix them by repairing systems.
So for every observation, ask:
What system does this belong to?
| Observation | Tag to System |
|---|---|
| “Failure to investigate OOS” | CAPA / Lab Investigations |
| “Inadequate cleaning validation” | Validation / Contamination Control |
| “Data not attributable” | Data Integrity / Digital Systems |
| “Batch records incomplete” | Documentation / QA Oversight |
Build this mapping into your response strategy.
Because FDA inspectors see through bandaids.
3. Use a Severity Matrix
Not all FDA 483s are created equal.
You need to know what to fix first.
Here’s a smart way to triage:
| Severity | Language Clues | What it Means |
|---|---|---|
| Critical | “Your firm failed to ensure…” | High-risk. Repeated. Expect Warning Letter if ignored. |
| Major | “Procedures were not followed…” | SOP exists but not implemented. Cultural or training issue. |
| Minor | “SOP does not adequately…” | Technical gap. Fixable with better documentation or controls. |
Start with Criticals.
They’re system-level breakdowns.
If you don’t fix those, nothing else matters.
4. Look for Patterns Across Sites or Inspections
Reading one FDA 483 is good. Reading ten is better.
Ask yourself:
- Is this issue recurring at other sites or functions?
- Are these symptoms of poor global QA governance?
- Do the same systems keep failing under pressure?
If you keep seeing the same CAPA failures, it’s a corporate one.
5. Don’t Treat FDA 483s Lightly
This is where 90% of pharma firms go wrong.
They treat each FDA 483 point like a to-do item:
- Update SOP
- Retrain operators
- Close CAPA
But that doesn’t fix the system. It just masks the outcome.
FDA expects you to answer:
“Why did this happen in the first place and what did you change to make sure it won’t happen again?”
That’s system-level thinking.
And that’s what gets you out of trouble for good.
Bottom Line:
A FDA 483 is a window into how FDA sees your quality culture.
- Read between the lines.
- Track the language.
- Map the systems.
- Prioritize the criticals.
Solve the root, not the symptom.
That’s how real QA leaders use FDA 483s to build bulletproof systems and not just pass audits.
Chapter 5: The 30-Day Golden Window
FDA gives you 15 business days to respond to a Form FDA 483.
But if you’re waiting till Day 14, you’re already behind.
The best firms start within 48 hours.
Why?
Because the clock is ticking and you are at the dead-end.
The Real Game: Show Control Fast
FDA doesn’t just read your response. They assess how fast, how serious, and how system-level your reaction is.
This is your one shot to say:
“We found it. We fixed it. We own it.”
And if you nail it, you stay out of a Warning Letter.
The Pro-Tier Response Checklist
If you’re serious about winning FDA’s trust, your FDA 483 response must check every box:
1. Acknowledge Every Observation
Don’t ignore or downplay.
FDA hates vague responses like:
- “We will consider revising the procedure…”
- “Operator retraining is planned…”
Instead, say:
“We acknowledge Observation 1 and have initiated immediate corrective and preventive actions. Root cause analysis is complete.”
Pro tip:
Never challenge the observation unless you have unshakable evidence. Even then, tread lightly. Arrogance invites escalation.
2. Attach Updated SOPs
Saying “we’ll revise the SOP” means nothing.
Attach the actual revised SOP version-controlled, QA-approved, and ready for implementation.
Even better, highlight key changes in the document.
Make it easy for the reviewer to see what’s improved.
3. Split CAPAs: Immediate + Long-Term
FDA wants to see both:
- Short-term action: What did you fix immediately?
- Systemic prevention: How are you stopping this from happening again?
Example:
- Immediate: Discarded affected batches, retrained line operators.
- Long-term: Redesigned equipment cleaning validation protocol with new acceptance criteria.
That’s control.
4. Assign a Responsible Person + Due Dates
FDA wants accountability.
Every CAPA must have:
- A single owner
- A realistic target completion date
- A verification step
Use a table like this in your response:
| CAPA ID | Action | Owner | Due Date | Verification Method |
|---|---|---|---|---|
| CAPA-001 | Revise SOP | QA Lead | July 10 | QA Approval Record |
| CAPA-002 | Train Staff | Training Head | July 15 | Attendance Log + Quiz |
Simple. Clear. Auditable.
5. Add Training Records
If your fix involves people, show proof they were trained.
Don’t just say “Training conducted.”
Attach:
- Signed attendance logs
- Training material used
- Quiz or assessment scores
Bonus points if you tie it back to SOP revision dates.
6. Include Evidence of Verification Steps
This is where most firms get lazy and where FDA turns skeptical.
Example:
“We revised the cleaning SOP.”
Great. But did you verify it works?
Add:
- Swab recovery test results
- Revised visual inspection criteria
- Mock batch run with enhanced sampling
FDA loves evidence of follow-through.
Because it proves the fix isn’t just theoretical but it’s working.
Pro Tip: Your Tone Matters
FDA isn’t just looking for technical fixes.
They’re reading your culture.
So avoid ❌ :
- Defensive language
- Passive voice
- Over-promising
Instead, use ✅:
- Assertive, respectful tone
- Clear commitments
- Transparent timelines
Remember: You’re fixing a finding and restoring the trust.
Bottom Line:
FDA 483s are a list of violations and a test of your maturity.
You have 15 days but only one reputation.
Use the window wisely.
Respond like a leader, not a regulator chaser.
And remember:
Speed + Systemic Fixes = No Warning Letter
That’s how elite pharma companies protect licenses, products, and lives.
Chapter 6: Preparing for the Next FDA Inspection,
5-Step Readiness Blueprint
When the FDA knocks, you don’t get a second chance to “get ready.”
There’s only one real strategy: stay ready.
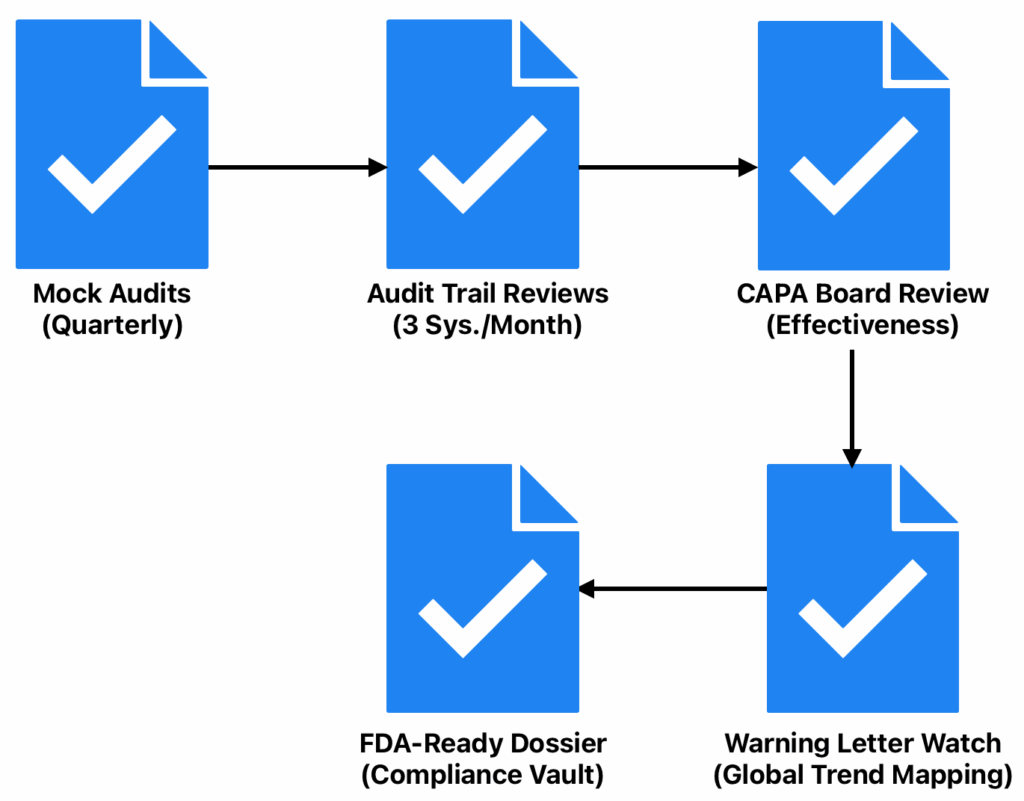
Here’s your 5-Step Inspection Readiness Blueprint designed for overseas pharma sites (OUS) that want to flip the script from reactive panic to confident control.
1. Mock Audits (Quarterly)
Think you’re ready? Prove it.
Bring in independent SMEs or ex-FDA inspectors every quarter. Not internal QA staff.
You need fresh eyes who aren’t blind to site routines or politics.
What they uncover might sting, but it’s better to bleed in a drill than hemorrhage during the real thing.
Pro Tip:
Alternate focus:
- Quarter 1: Data Integrity Deep Dive
- Quarter 2: Process Validation and Cleaning Validation Stress Test
- Quarter 3: Quality Unit Oversight Walkthrough
- Quarter 4: Full-system Simulated FDA Audit
This keeps readiness dynamic.
2. Audit Trail Reviews (3 Systems/Month)
Audit trails are the forensic backbone of electronic compliance.
Yet most firms never touch them unless auditors ask.
Here’s your fix:
Every month, randomly select 3 computerized systems, one from each critical zone:
- CSV system (e.g., LIMS, TrackWise)
- QC instrument (e.g., HPLC, NMR)
- Production equipment (e.g., SCADA, DCS)
Then review:
- Who made changes?
- What was changed?
- When?
- Why?
- Was it authorized?
Bonus:
Document your review. Not just for readiness but as a deterrent signal to potential data manipulation internally.
3. CAPA Board Review (With Effectiveness Checks)
Most sites track CAPAs like a to-do list. That’s not enough.
Your CAPA board needs 2 layers:
- Open CAPAs with owners, root causes, and due dates
- Effectiveness Reviews as a proof that fixes worked in the real world
FDA inspectors now ask:
“How do you know the problem won’t return?”
If you can’t prove that, you’ve failed no matter how many CAPAs you closed.
Monthly Review Meeting Tip:
Assign QA leads to test a random 10% of “closed” CAPAs for sustained effectiveness.
4. Warning Letter Watch (Global Trend Mapping)
Don’t wait for your own Form 483.
Learn from your peer’s mistakes especially those in similar markets, tech platforms, or countries.
Start a simple Warning Letter Watch Program:
- Every week, assign a team member to scan the FDA website
- Create a running tracker of major observations
- Tag them by system (e.g., “Data Integrity – Lab” or “Cleaning – Equipment A”)
- Score your own site against them quarterly
Why it works:
FDA doesn’t change its themes every quarter, it rotates them across regions.
What hit a Malaysian plant last month might hit you next.
5. FDA-Ready Dossier (Your One-Click Compliance Vault)
Let’s be honest: Most firms fumble when asked:
“Can you show us your master plan, last 3 years of deviations, and cleaning validation report for Line B?”
Cue the scramble. The missing file. The wrong version. Or worse… no document at all.
Fix it with an FDA-Ready Dossier, a digital (or cloud) folder that holds:
- Master Validation Plans
- Cleaning Validation Reports
- Deviation & CAPA Logs
- Data Integrity Policy
- Training Records
- Key SOPs (controlled versions only)
Bonus Points:
- Keep it on a secure, read-only platform
- Grant “View Access” only to specific QA staff
- Refresh every 30 days, minimum
Bottom Line:
Broken firms fear the FDA.
Smart firms prepare for it.
Elite firms are ready because their house is always in order.
Which one are you?
Chapter 7: Case Studies That Changed the Game
When it comes to FDA compliance, you want real stories. Real stakes. Real outcomes.
These two overseas pharma sites, one in Brazil, one in India; faced serious regulatory fire. But they didn’t fold. They acted fast, and smart.
Here’s what happened.
Case 1: Brazilian Sterile Facility
The Crime:
Environmental monitoring data was falsified.
Operators were backdating entries, “filling in” data that didn’t exist, and masking failed results.
The lab manager thought no one would notice.
The FDA did.
The Inspection Outcome:
- 3 major observations under 21 CFR Part 211.192 and 211.198
- Multiple data integrity breaches
- Risk to sterility assurance
What They Did Next:
This site didn’t waste time blaming. They went straight to data governance triage:
- Decommissioned the manual EM logbook system
- Migrated to a validated, Part 11-compliant EM software
- Rolled out 21 CFR Part 11 and ALCOA+ training to all QC and QA staff
- Appointed a Data Governance Officer
- Conducted forensic review of 6 months of EM entries
- Self-reported their findings, proactively
Outcome:
- FDA issued a Warning Letter
- But the site responded in less than 10 days
- Delivered a full remediation plan, with timelines and training logs
- FDA closed the WL in just 9 months, a rare success
Key Takeaway:
Data falsification isn’t a death sentence if your remediation is swift, deep, and backed by proof.
Case 2: Indian Oral Solid Dosage (OSD) Site
The Crime:
- Cross-contamination between product lines
- Cleaning validation failed to detect high-risk carryover
- Swabs showed active ingredient residues 800x above the limit
- Worst part? The same CV protocol had been used for 11 product campaigns
What the FDA Said:
The inspection team flagged:
- Inadequate CV protocol
- No justification for acceptance criteria
- “Visually clean” was used as a substitute for validated limits
- No periodic re-validation
This was textbook 21 CFR Part 211.67(b) and 211.160(b) violations.
The Turning Point:
Before waiting for the 483, the site’s Quality Head acted:
- Paused all manufacturing
- Commissioned an external Cleaning Validation consultant
- Rewrote the Cleaning Validation protocol from scratch with matrix grouping logic
- Repeated CV studies on all 11 products
- Retrained 40 cleaning operators and QA reviewers
- Created a digital CV master file: traceable, version-controlled
Outcome:
- Issued FDA 483
- But acknowledged the firm’s “exceptional post-inspection response”
- No Warning Letter was issued
- Site resumed commercial production within 3 months
Key Takeaway:
Your response speed and technical depth can flip the outcome even if the inspection outcome looks bad initially.
Bottom Line:
Both companies made mistakes.
Big ones.
But instead of denial or delay, they leaned into the problem and treated compliance like crisis management.
What set them apart?
- Immediate containment
- Honest gap assessment
- Visible executive leadership
- Bulletproof documentation
- And follow-through
That’s how you win back the FDA’s trust.
The question is:
If the same thing happened at your site tomorrow, would you be ready to respond this way?
Chapter 8: Future-Proofing Your Compliance System
Fixing today’s gaps won’t save you tomorrow.
FDA expectations are not standing still. Neither should your compliance strategy.
If you’re only reacting to FDA 483s, you’re already behind.
To win this game long-term, you need to build compliance into your company’s DNA.
That’s where most overseas pharma sites go wrong.
They think:
- SOP = Control
- Audit = Event
- Training = Attendance
That mindset leads to warning letters.
Instead, flip the script.
Here’s a 3-move framework to future-proof your operations – even if the FDA walks in unannounced next Monday.
Move #1: Digitalize Everything That Matters
Paper SOPs? Excel deviation logs? Email-based CAPAs?
That won’t cut it anymore.
What the FDA expects now:
- Validated QMS platforms (trackwise, Veeva, MasterControl)
- Part 11-compliant Audit Trail for every change, every record
- LMS (Learning Management Systems) that track real-time role-based training
💡 Real Tip: When choosing software, make sure it’s:
- Validated under GAMP 5 and followed 21 CFR Part 11
- Supports electronic signatures
- Can generate complete, secure, and enduring audit trails
This isn’t about digitization for show, it’s compliance-grade digitalization.
Move #2: Train for Insight, Not Just SOPs
Most firms treat training casually:
- Attendance sheet? ✔️
- PowerPoint review? ✔️
- Quiz? Maybe.
That’s not training. That’s liability.
You want your teams to think like FDA investigators.
Teach them:
- How to spot red flags in records
- How to defend procedures with evidence
- How to connect their task to 21 CFR requirements
Example:
Instead of just reading the cleaning SOP, ask:
- Can you explain how this aligns with 211.67(b)?
- What would you show an inspector if they asked about Cleaning Validation for this equipment?
That’s compliance intelligence and it only happens when you elevate training from “read and sign” to “learn and apply”.
Move #3: Benchmark in Real Time
The FDA is evolving.
If you’re not tracking FDA 483 trends, Warning Letter patterns, and global inspection outcomes, you’re flying blind.
Top-tier sites are already using:
- Redica Systems
- FDAzilla
- And soon PharmaGxP’s Audit Tracker
Imagine this:
You’re a QA head in Hyderabad.
Get a FDA 483 on a facility in Malaysia for OOS handling.
You get the alert.
You review your own OOS SOPs the next day.
That’s not just proactive. That’s regulatory intelligence in action.
Your competitors will be reacting.
You’ll be preventing.
Bottom Line:
Don’t treat compliance like a repair job.
Treat it like a system build.
Digital platforms, insight-driven training, and intelligence tools are the baseline for any serious pharma company targeting U.S. markets.
Because the next audit is both: a risk and your reputation test.
Conclusion:
From Survival Mode to Strategic Compliance
Let’s cut to the chase.
Compliance is your license to operate.
And the FDA isn’t pulling punches anymore. They’re looking for systems.
So, here’s what the best in class are doing:
- Embedding ALCOA+ into every data-generating process
- Validating cleaning with risk-based science, not legacy SOPs
- Empowering the Quality Unit with final say, not just a seat at the table
- Training teams to think like auditors, not just follow routine tasks
But here’s the part most companies still miss:
You can’t audit your way out of a weak culture.
If quality is something you prepare for before inspections, you’re already behind.
Top-performing sites?
Quality is how they operate every day.
It’s in
- How they review batch records.
- How they log a deviation.
- How they handle the unexpected without fear or shortcuts.
That mindset doesn’t just avoid FDA 483s. It builds trust with regulators, customers, and the patients you serve.
So here’s your call to action:
What’s the #1 challenge your team faces in building a truly inspection-ready QMS?
Drop it in the comments and let’s solve it together.
Because is your competitive edge. Stop treating it like a department. It is everyone’s responsibility.
References:
- FDA Announces Expanded Use of Unannounced Inspections at Foreign Manufacturing Facilities
- FDA 483 Trends – Regulatory Compliance Associates, accessed June 18, 2025, https://www.rcainc.com/published-articles/fda-483-trends/
- 2024 Trends In FDA Observations For Sterile Drug Manufacturers – Pharmaceutical Online, accessed June 18, 2025, https://www.pharmaceuticalonline.com/doc/2024-trends-in-fda-observations-for-sterile-drug-manufacturers-0001
- Best Practices in Cleaning Validation – Rephine
- FDA Inspection Observations Database – FDA.gov
- https://www.fda.gov/inspections-compliance