Preparative HPLC System: A Definitive Guide to RP-HPLC

Based on their application, HPLC systems are basically of two types.
- Preparative
- Analytical
Preparative HPLC systems are mainly used to isolate, purify, and deliver the protein from a solution mixture.
HPLC means High-Performance Liquid Chromatography OR High-Pressure Liquid Chromatography.
In this post, we’ll discuss about the design aspects of the Reversed Phase (RP) Preparative HPLC columns (RP-HPLC).
The separation mechanism in RP-HPLC systems depends on the interaction between mobile and stationary phase. Typically, solvent in the mobile phase and hydrophobic resin in the stationary phase.
Table of Content
Difference between Analytical and Preparative HPLC
Preparative HPLC systems are developed and used for intermediate purification of biosimilars, APIs, and other smaller molecules. While analytical ones have different purpose.
To make it easy, here’s a quick take.
| Analytical HPLC | Preparative HPLC |
|---|---|
| Used in analysis of target compounds | Used in purification of target compound analyzed in analytical HPLC |
| Tells us about the quantity and nature of the component | Separates and collects components of interest in the required quantity |
| They are lab-scale | They are process-scale |
| Have a small column size (column diameter) | Have large column sizes (column diameter) |
| Deals with small particle sizes of stationary resins | Deals with large particle sizes of stationary resins |
| Exerts high back-pressure due to compact size | Back-pressure is somewhat low due to the large scale dimensions of the column |
| Operated at lower flowrates ranging from 0.006 to 0.6 ml/min | Operated at a high flow rate of 0.6 to 12 L/min |
| Stand-alone technology | Technology stands on analytical HPLC |
Column Preparation Process
Using a moving piston, the column design facilitates the principle of Dynamic Axial Compression (DAC). The use of the piston delivers dynamic compression helpful in column packing, unpacking, and maintaining packing pressure. However, different suppliers may adopt different operational mechanisms.
Column Preparation – Working Principle
For more clarity, see the below visualization.
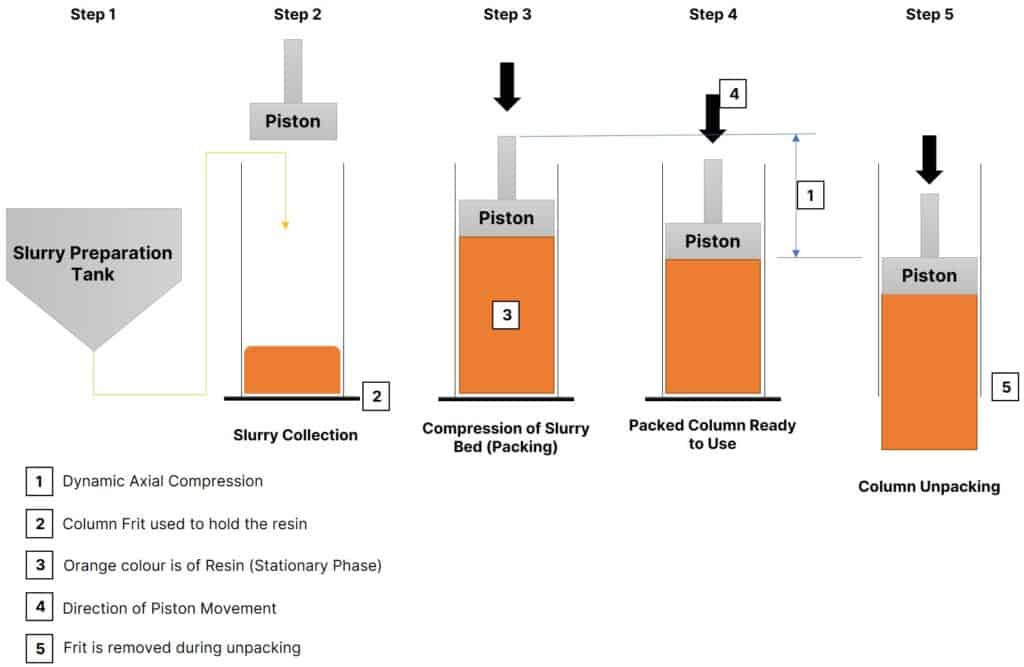
When resin is the packing material and a stationary phase, the column preparation process somehow goes like this:
- Resin first undergoes slurry preparation in a suitable process vessel.
- The slurry then gets transferred to the column by the application of pressure or a suitable pump.
- After the slurry transfer completes, the piston starts to compress the slurry at two different levels.
- Air present in the column space purges out through the piston.
- The solvent in the packing material leaves through the bottom flange and in return, packing material experiences compression.
- Column packing is complete. Now, the column is ready for batch process.
- Piston pressure is maintained on the packed bed using Dynamic Axial Compression. This is called packing pressure that helps avoid the potential of building vacant spaces inside the bed.
After the resin is completely used, the column is unpacked by lowering the piston and removing the bottom flange and frit.
The important physical parameters of the column design are:
- Column Internal Diameter (mm)
- Stroke of the Piston (mm)
- Maximum Working Pressure in bar(g)
- Maximum Bed Height (mm)
- Bed Volume (L)
Column Specification Terminology
Typically, the reversed phase HPLC columns are specified as below.
Specification of the Column: C8, 50x1200mm, 7µm, 100Å
- The first one, C8 is a chain length of carbon in the stationary phase i.e. resin.
- The second one is the column dimension in terms of length and internal diameter.
- Third, the particle size of the resin.
- Fourth, the pore size of the resin.
Particle size for silica-based carbon chain resins generally ranges between 3 to 15µm. Also, there are different carbon chain lengths ranging from C4 to C18.
These resins come with their own mechanical stability with different maximum packing pressure they can sustain.
Following are the parameters looked into while selecting the resin:
- Particle Size and Distribution
- Pore Size and Distribution
- Specific Surface Area
- Pore Volume
- Chemical Purity and Stability
- Packing Density
- Coverage etc.
Phases of HPLC System
HPLC systems have two phases.
- Stationary Phase
- Mobile Phase
Difference Between Normal and Reversed Phase
Two types of HPLC systems based on their phase are:
- Normal-Phase (NP-HPLC)
- Reversed-Phase (RP-HPLC)
Here’s a list of quick and non-exhaustive differences.
| Normal-Phase (NP-HPLC) | Reversed Phase (RP-HPLC) |
|---|---|
| Superseded by RP-HPLC due to inconsistent reproducibility of retention times | Advancement in NP-HPLC that gives better control over retention time reproducibility |
| Utilize the polar stationary phase | Utilize a non-polar stationary phase |
| Utilize a less or non-polar non-aqueous mobile phase | Utilize a polar aqueous mobile phase |
| Only organic solvents are used | Solvents like Methanol and Acetonitrile are used |
| The rate of elution is slow in polar molecules and vice-versa | The rate of elution is rapid in polar molecules and vice-versa |
| Analyte elution depends on the increasing polarity of the mobile phase | Analyte elution depends on the decreasing polarity of the mobile phase |
| In short, 1. Stationary Phase is Polar (Pure Silica) 2. Mobile Phase is Non-Polar (Non-Aqueous Solvents) | In short, 1. Stationary Phase is Non-Polar (Advanced Silica with a modified surface) 2. Mobile Phase is Polar (Aqueous Solvents) |
Components of Column Module
These RP-HPLC systems have a very complex design on a preparative scale and are hence generally classified in different sections.
- Column – Main working frame of HPLC system where separation happens.
- Column Skid – Downstream of the column that handles different components including UV detection lamps, three-way valve assemblies, and related sensors.
- Gradient Pump Skid – These are very heavy-duty pumps required to bring out the component of interest from the chromatographic bed.
- Remote Control – Basically either HMI or SCADA system to monitor, operate and control the process remotely.
- Hydraulic System – Used to move the jack in up and down direction during packing and unpacking operations respectively.
These are just the distinctions of critical areas of the HPLC system that again contain additional components explained below.
Other Components
Bottom Frit
It is used to close the bottom of the column with a suitable seal and flange arrangement. The frits are used to provide proper liquid distribution over the cross-section of the column through the porous and metallic structures. This in turn helps in achieving consistent and better asymmetry profiles during elution.
Piston
The piston is important because it performs three key operations in the column
- Packing
- Unpacking
- Maintain column pressure dynamically while in use
Piston Seal, Compression and Hydraulics
These altogether ensure the complete tightness between the piston and column wall with the help of a compression system. Apart from the above, the Hydraulic unit performs the following critical functions.
- Controls the piston movement
- Maintains the dynamic compression pressure on the resin bed
Pumps
When the preparative HPLC systems perform neat and clean, pumping systems are the backbones of it. Different applications require different pumping systems.
For example, a pneumatic pump would serve the purpose of dynamic compression on the resin bed and help maintain the jack’s speed movement. An electrical pump would serve the purpose of piston displacement speed during column packing. While the gradient pump would only be concerned about handling the flow rate during the process cycle.
Other Instrumentation
Apart from the above critical components, preparative HPLC systems require various supporting components and associated instrumentation.
This includes an oil tank to store hydraulic oil, pressure gauges, safety relief valves, pneumatic distributors, air pressure regulators, pneumatically actuated valves, valve gangs, limit switches, and a control panel to operate the equipment with ease.
Compression Ratio
There is sequential proportionality between the following.
- Chromatographic Bed Pressure (As applied by Piston)
- Oil Pressure (Associated with the jack)
- Air pressure under dynamic compression
The compression ratio helps to calculate the amount of piston pressure to be applied to maintain dynamic compression during a column operation. In simple form, piston pressure increases linearly with the oil pressure. As a matter of calculation, this ratio should be approx. three (3).
Compression Ratio = (Hydraulic Pressure) / (Piston Pressure) = ~ 3
Based on this, we can say, the hydraulic pressure of 300 bar(g) results in a piston pressure of 100 bar(g).
Slurry Preparation for Packing
The quantity of packing material that should be used in the slurry preparation depends on the following things:
- Bed Height
- Bed Density (Directly proportional to packing pressure)
- Packing Pressure
- Bed Permeability
- Choosing the right solvent for the right phase
- Less viscous slurry
Along with this, it is equally important to discourage the use of magnetic stirrers due to chances of particle abrasion. Alternatively, a recirculation pump will help to form a uniform mixture.
For that, packing material should be poured into the solvent instead of solvent in the packing material. Once the slurry is prepared, it is then transferred into the column appropriately.
Buffer Preparation for RP-HPLC
These are the chemical compositions of the solvent that flow through the column bed of resin helping to bind and elute the component of interest at a particular time.
Apart from that, buffers are adjusted to the compatible pH value and added with salts for better column performance.
Generally, there are 2 or 3 different compositions of the aqueous solvent such as Acetonitrile that are used for different physicochemical interactions we’ll see below.
Process Flow of Reversed Phase Preparative HPLC Systems
The process of conducting the column operation for the commercial separation of proteins is pretty straightforward.
It works on the principle of Adsorption and Desorption. A compound of interest acts as an adsorbate while the resin acts as an adsorbent.
Depending on the nature of the desired component, there exist the following set of operations.
- Equilibration – Performed with less solvent composition buffer to improve the ionic strength of the protein in easily binding the resin.
- Loading – Component of interest preferably in liquid form that binds to the resin.
- Gradient Elution– The main phase that’ll trigger the elution by dynamically increasing the proportion of the solvent composition.
- Buffer Wash – Moderate solvent composition to remove the impurities associated with the product from the resin bed.
- Column Regeneration – A wash that removes any deposits on the surface of the resin, including mobile phase contaminants.
The below diagram outlines the overall process scheme of an RP-HPLC system.
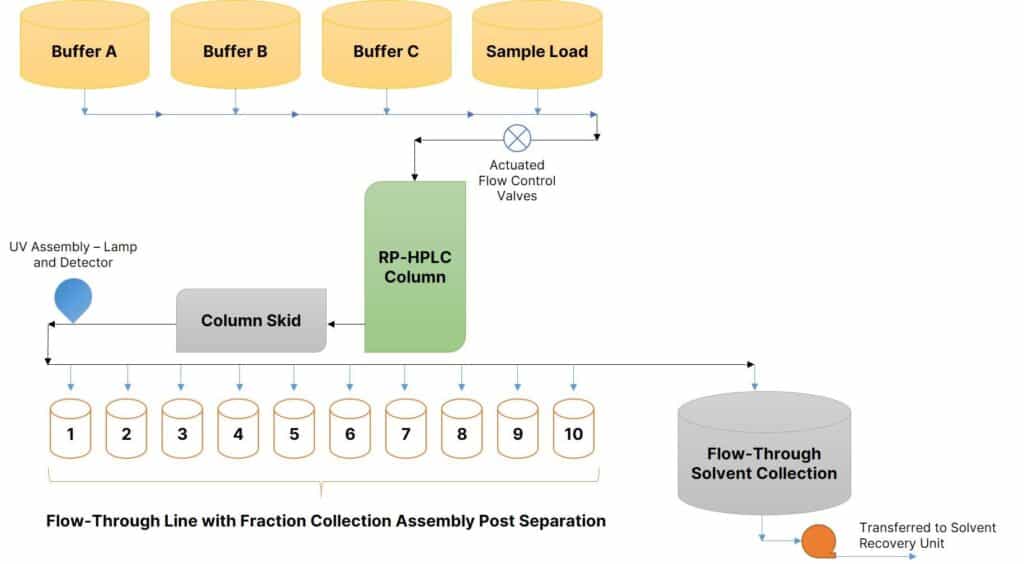
This complete process is dependent on the chromatograph running in real-time typically on a SCADA system backed by a UV assembly. Let’s move on step-by-step.
1. Column Equilibration – Gives Ionic Strength to Protein
Almost all preparative column resins are equilibrated with a buffer compatible with the compound of interest. These buffers are passed through the resin bed.
The amount of the equilibration buffer to be passed is approx. considered in the range of 5 to 10 times the column bed volume.
Column bed volume can be calculated by using the volume of cylinder formula i.e.
Bed Volume in L = (π * (Bed Height in mm) * (Internal Radius in mm)2 * 10-6) in L
For example, for a bed height of 500mm and column internal radius of 300mm,
Bed Volume = 3.14 * 500 * (300)2 * 10-6 = ~ 141 L
This means the quantity of equilibration buffer should be in the range of 700 to 1400 L for a bed volume of 141 L.
Bed height and resin quantity are decided based on the amount of sample that is about to bind the resin. This in turn helps in bed volume calculations.
2. Loading – Binds the Protein
Once the column is equilibrated, the sample load is loaded onto the column bed. Then, the resin and protein interact. The protein binds to the resin particles through adsorption.
It is recommended to control optimum flow rate during loading because a higher side flow rate would not allow sufficient time for binding or simply overload the column and a lower side flow rate would unnecessarily increase the time of the loading cycle.
A dedicated pump installed prior to the column is generally used to carry out loading operations. The volume of the load depends on the binding capacity of the resin.
Suppose if the resin is able to bind a maximum of 500gms of the load, then loading a complete load volume that contains 1500gms would be of no sense. Rather, it’d be great to divide that into 3 to 4 lots. Therefore, quantification of the load prior to loading becomes important to overcome the overloading issues.
3. Gradient Phase – Where Elution Occurs
Once the loading step completes, the gradient phase starts. This is the step that separates the compound of interest with high purity. Meaning, most of the weak proteins and impurities are washed off from the resin.
In the gradient phase, the resin is exposed to the increasing proportion of buffer B over a period of time. It is important to note, the gradient is always started with a small % of buffer B preferably less than 10%.
Starting with 0% buffer B may directly impact the chromatographic separation as the binding ability of the protein may deteriorate.
Though the proteins strongly adsorb on the surface of resin particles, they are very prone to leave the surface at higher polar concentrations. The polarity of the aqueous mobile phase depends on the composition of the solvent added to it. As the composition increases, polarity also increases.
The overall operating range for the gradient phase varies from 100% of buffer A to 100% of buffer B dynamically.
To quantify the separation, calculating Retention Time or Rs Value (Resolution) is the simplest method measured as the volume per average width of the respective peak.
However, there are different types of elution profiles either employed independently or combinedly.
Isocratic Elution vs. Gradient Elution
When the composition of the mobile phase remains constant and polar throughout the separation, it is called Isocratic Elution.
While in the Gradient Elution, the composition changes from high polar to low polar. When dealing with protein purifications, gradient gives more clarity in differentiating various peaks without affecting the resolution. When choosing the isocratic elutions, it is important to note; they suffer in detectability in the long run due to band broadening.
This does not mean one should only use either of the ones. The selection of the elution can be determined on the basis of accurate concentrations. The following diagram shows the difference between them.
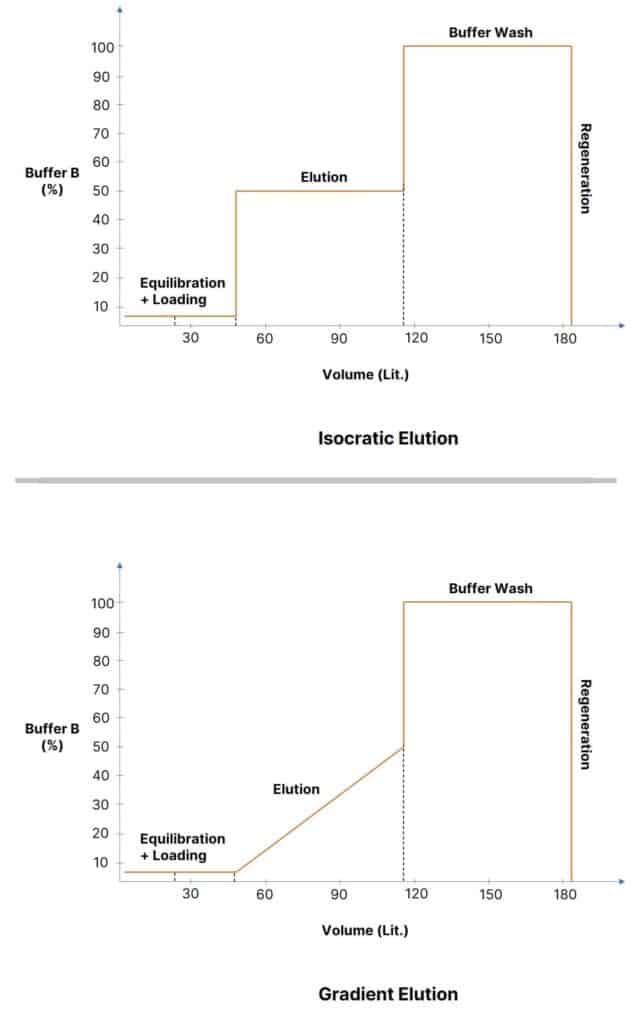
There are two ways to design the gradient for reversed phase preparative HPLC systems.
Step Gradient
It is a series of isocratic elution carried out step-wise at different buffer B compositions. If the concentration at which the compound of interest leaves the resin is known, a step-by-step isocratic elution approach saves time in future separations rather than following gradient elution on a regular basis.
Step gradient method uses simple instrumentation and hence it is preferred for a process whose:
- Desired resolution can be obtained.
- Separations are carried out at low regime resolutions.
Continuous Linear Gradient
When dealing with a larger regime of resolutions, the continuous gradient is the choice of separation. In this type, the composition of the mobile phase changes linearly with time.
Unlike isocratic elution, this gradient keeps band broadening to a minimum and hence helps in higher detectability.
4. Buffer Wash – Separates Other Associated Impurities
A buffer wash of 100% composition should flow through the column bed upon completing the gradient phase.
This helps in removing any protein traces that may have been stuck on the resin and associated impurities.
Generally, the sudden change in mobile phase composition may damage the packing quality and hence this wash helps in avoiding that. This step will also help the column to set it up for further use.
5. Regeneration – Helps in Removing Deposits
After washing, regenerating the resin bed for the next run is critical and requires a higher composition of the aqueous polar solvent, suitably a separate buffer C.
If this step is not performed, the issue would be catastrophic in terms of column performance including:
- A sudden increase in back-pressure
- Changes in retention times
- Poor separation
- Poor peak broadening
These signs indicate that the resin has been impacted by the compound deposits or precipitations on its surface. Flow should be on the lower side with up to 10 column volumes.
A proper regeneration technique should be applied either in the normal flow or reversed flow without affecting the column performance.
Remember, back pressure should not be more than packing pressure. This ensures the column is not clogged and can be used for further separations. If backpressure > packing pressure, the resin should be washed with acid in an upward direction ensuring complete residue desorption from the resin surface. Additionally, filters to be replaced if chocked for both load samples and mobile phases. Don’t forget to test HETP after column wash.
Reversed phase i.e., stationary phase resins should be stored in a mixture of water and solvent. Care should be taken to avoid using any additives that may precipitate when stored.
When the operation of the column is complete, it is generally washed for 10 to 15 minutes in proportional composition.
Now that you’ve seen the process steps for reversed phase preparative HPLC systems, let us see an example of a typical chromatogram obtained after completion of the recipe execution.
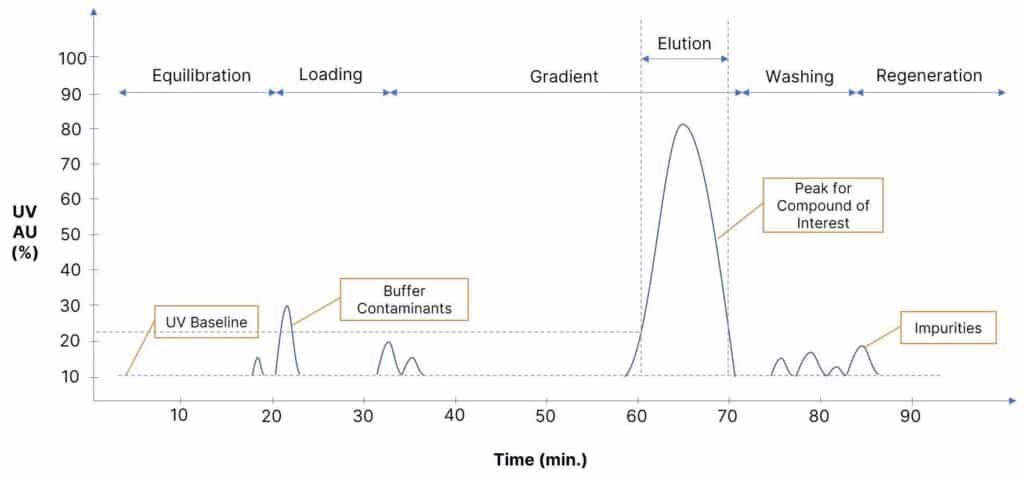
Performance Indicators
The chromatogram runs in real-time during the complete operation of the process.
Let us understand a few important indicators and factors responsible for effective column performance.
Critical Process Parameters:
When it comes to protein separation by reversed phase chromatography, it is important to understand that small fluctuation in any of the process parameters has a critical impact on product quality.
It may ultimately denature the protein of interest due to its sensitive nature. The following are the commonly considered critical process parameters for preparative HPLC systems.
- Mobile Phase: Choose the correct organic modifier such as Acetonitrile which is UV transparent.
- Temperature: Increase in temperature will cause a decrease in the viscosity of the mobile phase. This also affects the compression ratio due to changes in oil properties.
- Flow rate: Affects dynamic binding capacity (resin binding kinetics) especially during loading of process scale preparative HPLC systems.
- Gradient-Elution profile: Typically a linear gradient method is preferred for preparative HPLC systems. Meaning, the gradient flow will range from higher to lower polarity levels over the period of time.
- Stationary and Mobile phase physicochemical interactions.
Evaluating Column Efficiency (Half-Height Method):
The efficiency of the column is driven by Height Equivalent to Theoretical Plate (HETP) expressed in terms of the number of theoretical plates (N).
To evaluate the efficiency of preparative HPLC systems, this dimensionless number help us understand:
- Column packing is good or needs repacking
- Whether the packing material is intact
When dealing with protein manufacturing such as Insulin, the peak of the protein in the chromatogram follows the Gaussian path.
In such cases, a valid and most preferred way to estimate the column performance is by using the peak width at 50% peak height. Hence, this is called the half-height method.
Let’s plot a graph considering the time of injection is around 48 minutes. Just to show differently than our previous chromatogram where the time of injection was 20 minutes.
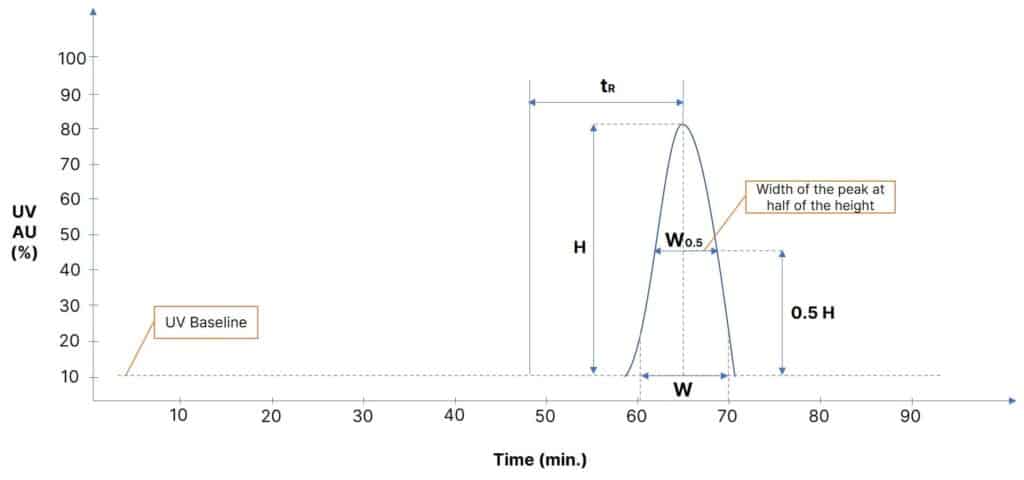
Column Efficiency for preparative HPLC systems is calculated in terms of the number of theoretical plates (N) per column as below.
N = 5.54 * (tR / W0.5)2
Once the number of theoretical plates per column is calculated, you may also calculate H. i.e., Height Equivalent to Theoretical Plate (HETP) which is the number of theoretical plates per meter of the given column length.
H = L / N
Abbreviations:
tR – Retention time
H – Height of the curve
W0.5 – Width of the peak at half-height
W – Width of the peak 10% above the baseline (just at the peak tailing)
L – Column Length
N – Number of theoretical plates per column
Though we’ve discussed the half-height method for evaluating column efficiency, there are other alternative methods available to derive the same such as:
- Tangent-Line Method → N = 16 * (tR / W)2
- Modified Gaussian Method → N = 41.7 * ((tR / W0.1)2/((b/a)+1.25)) where W0.1 – Width at 10% height.
- Area Height Method → N = 2π * (tR.H / A)2
Either way, these methods will result in slight variations in “N” depending upon the peak characteristics and shape.
However, the half-height method is often adopted for SCADA systems that evaluates column efficiency seamlessly.
Peak sharpness increases with the increasing number of theoretical plates (N) per column. This means, the more the number of theoretical plates, the more is column efficiency.
FAQs:
1. How to Optimize the Column Efficiency?
By altering Flow rate or solvent velocity calculated at reduced plate height → Particle Size of the resin → Length of the column (L).
2. What is the Desired Elution Pressure? How to Tackle Back Pressure Issues?
The best way to confirm that no overpressure issue with the column exists is through the theoretical estimation of the elution pressure. Certain pressure drop (ΔP) will surely be there in the column and it can be estimated as below.
ΔP (bar) = 10-6ULηK0 / dp2
Where,
dp → Particle diameter
U → Solvent Velocity (U = Column Length / migration time of an unretained marker)
L → Column Length
η → mobile phase viscosity
K0 → permeability constant generally considered as ~600 for spherical beads and ~1000 for non-spherical beads.
This equation is derived from Darcy’s law of pressure drop. If the pressure drop is significant, it might be the situation of deteriorated packing material. Though in such cases, expert opinions are advised.
Peak Asymmetry Factor (As):
One of the important factors in measuring column efficiency, As in terms of an offset, tells us the behavior of the peak symmetry.
As can be calculated by dividing the center of the peak into two parts equally at peak width (W).
Suppose the left part from the division is “a” and the right part is “b”. The Peak Asymmetry Factor can then be calculated as:
As = b/a
If = 1, the peak is perfectly symmetrical,
if <1, its a fronting peak, and
if >1, its a tailing peak.
Evaluating Packing Efficiency:
A poorly packed column can significantly drop the performance of preparative RP-HPLC systems. By measurement, one can evaluate whether the column has been packed well.
Van Jozef van Deemter, a Dutch physicist and engineer, related the HETP of the column to the linear velocity of the mobile phase through mass transfer, chemical engineering thermodynamics, and reaction engineering.
The equation he provided is called Van Deemter’s Equation and expressed as:
HETP = A + (B/u) + (u*C)
Where,
A – Eddy diffusion (which results in peak broadening)
B – Longitudinal diffusion coefficient
C – mass transfer between mobile and stationary phase
u – flow velocity
The last term in the equation when multiplied with Peclet Number is called Rodrigues equation. This is an extended version of Van Deemter’s equation that indicates the efficiency of the resin bed for large pore-sized permeable particles.
Just to give you an idea, when we talk about reversed phase preparative HPLC systems,
A < 1 → Well packed column
1 < A < 3 → Column packing can be accepted conditionally.
A > 3 → Bad quality of either the packing material or the packing itself.Similarly, C > 0.3 → Inadequate mass transfer between mobile and stationary phase.
These values should not be considered exact as they may vary based on application.
Relationship Between Linear Velocity and Flow Rate:
In preparative HPLC systems or any chromatographic system, standardizing the flow rate for different columns is expressed by using linear velocity (m/s or any suitable unit) as a multiplication factor.
Linear velocity is the speed at which the mobile phase passes through the column.
During process scale-ups or scale-downs, using the same flow rate for different internal diameter columns is not a practical solution.
So, linear velocity is used to draw an equivalence between the two columns. Instead of using the same flow rate, linear velocity is kept constant to adjust the flow rate for the new column dimensions proportionally.
The following formula is used to calculate the new flow rate expressed as the square of the ratio of the column IDs:
New Flow Rate (LPM) = Previous Flow Rate (LPM) * (New ID / Old ID)2
Examples:
- Suppose the flow rate for the previous column having an internal diameter of 800mm was 15 LPM. The changed column diameter is now 1200mm. According to the above formula:
New Flow Rate (LPM) = 15 * (1200/800)2 = 15 * 2.25 = 33.75 LPM. - Suppose the flow rate for the previous column having an internal diameter of 1200 mm was 33.75 LPM. The changed column diameter is now 800mm. According to the above formula:
New Flow Rate (LPM) = 33.75 * (800/1200)2 = 33.75 * 0.45 = 15 LPM.
Retention or Capacity Factor (Kc):
During protein purification, it is important to keep a record of retention factors of the different compounds including the one of interest.
If they are the same, impurities will also follow the compound of interest and would yield impure protein at the time of elution.
Also called the Capacity factor, the Retention factor is the measure of proportional time a load sample stays in the stationary phase rather than the mobile phase. It is different than the binding capacity. Just to simplify, let us take a look again.
The mass of the solute in a specified amount of stationary phase that is able to bind at specified conditions is called Binding Capacity. The measure of proportional time a solute spends in the stationary phase rather than the mobile phase is called Capacity Factor (Kc).
How Binding Capacity is Different Than Capacity Factor
For every peak that logs into the chromatogram, retention factor or capacity factor for a loading sample can be estimated by:
Kc = (No. of moles present in stationary phase / No. of moles present in mobile phase)
Selectivity (α):
The ratio of retention factors expressed in a particular mobile phase composition as:
α = kb / ka = Vb /Va
Where,
kb – retention factor of a compound that retains on the resin for a long time
ka – retention factor of a compound that easily leaves the resin
Vb – retention volume of a compound that retains on the resin for a long time
Va – retention volume of a compound that easily leaves the resin
It is equivalent to the relative retention of the solute peaks. In other words, selectivity is the ability of a reversed phase preparative HPLC system to separate two analytes from each other.
Resolution (Rs):
This is the function of selectivity, retention factor, and the number of theoretical plates (N). Resolution Rs defines the separation of chromatogram peaks which is the main purpose of chromatography.
Changes in resolution can occur due to properties of the resin when equilibrated, washed, flow rate, load volume, and buffer composition.
Distance between the centers of the two elution peaks in terms of Retention time per average peak width. This is measured by considering the contribution of selectivity, efficiency, and retention factor
The formula to calculate Resolution is as below.
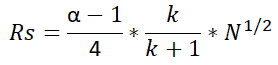
Where,
α – Selectivity (defined earlier)
k – retention factor
N – number of theoretical plates
Yield:
Yield is defined as the quantity of protein recovered with respect to the quantity of protein loaded. It is used to evaluate the productivity of preparative HPLC systems.
Yield (%) = (Quantity of Protein Recovered / Crude Protein Loaded) * 100
Common Problems and Troubleshooting
Poor Plate Counts:
Chromatograms result in poor plate counts due to overloading of the injection volume onto the column resin. General practice is that the injection volume should not be more than 10 to 15% of the loading flow rate.
This means sample quantities of 0.01 to 0.015 mg should be loaded per ml of column volume at max.
Another reason for poor plate count is dead space. Dead space is the overall volume occupied by injector assembly, tubing, UV detector assembly, and other mechanical components such as connectors, filters, etc.
Hence proper assembly of all the components plays a crucial role in maintaining sufficient plate counts.
Ghosting or Ghost Peaks:
Have you seen unknown peaks that neither resembles your compound of interest nor associated impurities? If so, it indicates poor quality mobile phase.
The organic impurities in the mobile phase may bind to the resin at significant levels such that during buffer wash after elution, these contaminants desorb from the surface of the resin and make an appearance in your chromatogram known as Ghost Peak.
However, they do not cause any harm in the short term but can severely damage the resin in the long term. Use a high-quality mobile phase and monitor regularly for such peaks.
Column Contamination:
It is the primary sign of partially chocked frits. This is caused when the loading sample and mobile phases are injected without using a filter that may contain certain physical components irrelevant to the desired separation.
Using proper filters preferably of small pore sizes would prevent the physical impurities from being injected onto the preparative HPLC systems.
Increased Backpressure:
Specifically, at the top of the bed, the chances of precipitation of compound of interest are high. In this scenario, it is important to clean the filters and the column. See FAQ-2 above for more details.
Poor Resolution:
As discussed earlier, poor resolution can be a result of poor selectivity. Sometimes, the reason might be poor column packing and column overloading.
Evaluating column efficiency and repacking the column if required, are the choices to overcome this problem. At the same time, cleaning and performing the regeneration step should be helpful.
Split Peaks:
When the elution strength of the injecting solvent is greater than the mobile phase, peaks can split or break because the two compounds may be separating almost at the same time.
To avoid this scenario, using a smaller resin particle size and greater column length will produce better separations.
Spikes In Chromatogram:
Spikes in the chromatogram show UV baseline rises from the initial value to the next value irrespective of changes in the concentration of the phase.
The main cause of this issue is related to the air bubbles trapped in the UV cell. Degassing UV assembly is the choice of treatment in such cases.
Periodic Column Cleaning:
Periodic column cleaning is recommended if the contaminants are column fouling. This is similar to what we’ve seen in the regeneration phase.
When the level of the contaminants is significant, a cleaning with low pH mobile phases is preferred.
Because, they wash off any contaminant on the reversed phase silica-based resins. Such resins contain Silanol groups that form gel aging when exposed for longer durations to aqueous mobile phases.
The general way of column cleaning is:
- Equilibrate the column by passing multiple column volumes of buffer A at a low flow rate with a minute proportion of any suitable acid in it. Sometimes, alkalies can also be considered.
- Run a dummy gradient starting from 100% buffer A and ending on 100% buffer B again containing a minute proportion of suitable acid. Remember to execute on approx. 25 column volumes.
- Perform a column equilibration similar to the above step, but this time starting from 100% buffer B to 100% buffer A.
- Finally, re-equilibrate the column using buffer A with multiple column volumes around 10x.
Automation
Custom-made automated software, preferably PLC and SCADA combinations, would help in delivering consistent and quality performance.
For a reliable process, the software may have following features:
- Robust control of Critical Process Parameters
- Column efficiency measurement tools
- Dynamic chromatograms with required filters
- Column Pressure and Temperature Indicators
- Automated Backflush Sequence Provision
- Alarms and Interlocks as per Process Flow
- Facilitating smooth fraction collection during elution
- Flow Totalizers to easily know how many column volumes we’re processing
- Hierarchy-based User Access Level Rights
- Compliant with 21 CFR Part 11 and GAMP5 Guidelines
While the list is limitative, using these automated features in your reversed phase preparative HPLC system can really strengthen the performance and regulatory compliance.
Conclusion
We’ve seen detailed aspects including column design, development, process application, and optimizing your preparative RP-HPLC systems from basics to troubleshooting.
When these systems are used in the downstream manufacturing of APIs, the collected fractions are further processed for polishing the proteins until they get lyophilized.
Reversed Phase HPLC is a very unique solution for protein purification in the downstream operations of the biosimilars.
Hope you find this guide useful.
It’ll be interesting to know, how you conduct your preparative HPLC system operations? What challenges do you face in implementing an effective protein purification? Let me know in the quick comment below.




I didn’t know that HPLC systems are made for intermediate purification. I had no idea that they could help with this. So I will try and find an HPLC system to help me out.
Hey Rick! Thanks for letting me know this article being useful for you. I would love to know what you’ve got. All the best!
Hi Saket, Good article on RP-Preparative HPLC.
Can you elaborate/help me in the Automation of fraction collection (for the compound of interest) process based on online UV signal?
Would be great if you can share specific information on this.
Thanks
Thanks, Mr. Naidu! Automated fraction collection based on an online UV signal is very sensitive and needs an in-depth retrospective analysis of the trend behaviors. I’d suggest you to drop an email at [email protected] and if you need a precise solution, let me take this forward systematically.